二聚超氧化物歧化酶的結構性變換和肌萎縮性側索硬化癥
- 2012-11-06
- 專題
1.肌萎縮側索硬化癥(ALS) 和突變型超氧化物歧化酶(SOD)
肌萎縮側索硬化癥(ALS)是一種漸進的麻痹性疾病,其特點是上下運動神經元選擇性退化。1,2)雖然ALS是一個顯著的偶發性疾病,但是~10%的案例是常染色體顯性遺傳(家族性ALS(fALS))和部分fALS案例是由SOD1基因突變導致的。3,4)SOD1產品基因,細胞質Cu,Zn-超氧化物歧化酶(SOD1),是一種廣泛表達酶,能夠催化超氧自由基歧化反應。5)
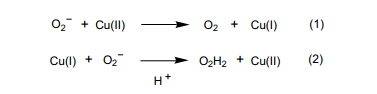
SOD(Cu/Zn)的晶體結構已經確認,8) Fig.1 (PDB, 1spd_x)所示它的二聚體結構。銅和鋅通過組氨酸咪唑環陰離子的連接在一起。
一些數據表明:SOD1突變的結果除了功能缺失導致肌萎縮側索硬化癥(ALS)以外,還有另外的結果。例如,一些fALS-相關的突變體SOD1s保留了完整的酶活性。6)此外,SOD1能夠導致小鼠失去知覺,不顯示ALS癥狀,然而轉基因小鼠表現出fALS-相關突變體G93A SOD1延伸性ALS-類似癥狀盡管也有內源性小鼠SOD1表現。7)最終在這個轉基因小鼠試驗中過表達人類野生型SOD1的失敗用于緩解ALS癥狀。7)
一種假設解釋說SOD1功能性的收獲是突變體的錯折疊改變了其催化機制,從而允許了過氧亞硝酸鹽9)和有可能的過氧化氫生成。10)另外一個重要的假說認為這個毒性源于SOD1細胞內聚集體。SOD1包涵體,與anti-抗體反應,是ALS 患者常見的運動神經元和附近的星形膠質細胞供給體。11)
然而,當我們考慮到蛋白質氧化鈣性有助于聚集體和蛋白酶抵抗力的時候,這兩種假設也不是相互排斥的。蛋白質聚集是很多種神經性失常變性疾病的病理性特征,12)包括Huntington’s,Alzheimer’s和 Parkinson’s等疾病。每一種情況下,SOD1s 突變體錯折疊和聚合傾向的可能性原理通過超過100不同的突變導致一個常見的表現型。盡管SOD1聚集體可能有的內在毒性或者是通過螯合分子伴侶導致的運動神經元毒性阻礙了蛋白酶的正常運作,但是SOD1聚集體的毒性起源尚未明確。
要了解ALS的發病機制,我們必須弄清楚如何改變SOD分子能誘導分子的損傷。為了進行這樣的調查,我們已經開始弄清楚了功能增益的原因和SOD聚集體在溶液中的機制。
SOD1的反應機制已經被許多學者進行了研究。最近Nishida等人對于這種酶基于通過使用模型化合物所得的結果有了一個新的假設機制。13)在上述第二步驟中 (2)我們已經指出了Cu--OOH形式的中間體形成的重要性(見圖表-I),這種過氧化氫能夠從野生型酶中立即祛除,因為過氧化氫和copper(II)離子與周圍的有機基團分子間作用基本上可以忽略不計。

Siddique等人7)已經確定了人類SOD的晶體結構,還有另外兩種SOD結構,并且已經確定fALS突變不能改變任何活性點的殘基包括基質中的靜電識別,金屬離子的結扎或者是活性點通道的形成,但是能夠檢測到銅離子(Ⅱ)周圍細微的變化。基于準確研究典型結構和蛋白質中疏水材料的積極影響的基礎上,6個fALS突變體有可能破壞亞單位折疊或者二聚體結構。但是,我并不認為這些對亞單位折疊或者二聚體結構的破壞性影響可以足夠的解釋所有fALS 發病原因。
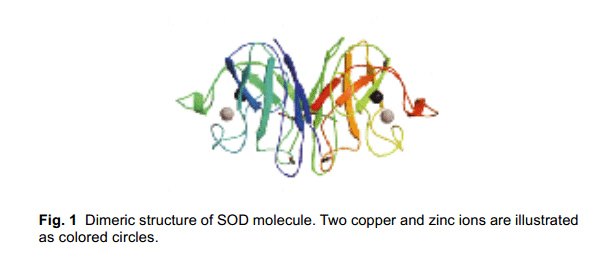
1997年, Yim等人報道了一個fALS 突變體(Gly93Ala=G93A)表現增強自由基生成活性的作用, 而它的歧化活性與野生型酶一樣。14)圖2為含有H2O2和人SOD 酶溶液中DMPO-OH radical加成物生成物ESR 波譜。據他們報道,突變體中自由基生成的活性,在低濃度H2O2條件下通過自旋捕捉法,相對于wild-type和G93A活性是增強的,wild-type<G93A<A4V,但是上述觀測到的原因還沒有詳盡的描述。
2. “增益功能”SOD酶起源與Cu(II)-過氧化氫加合物獨特的反應性能
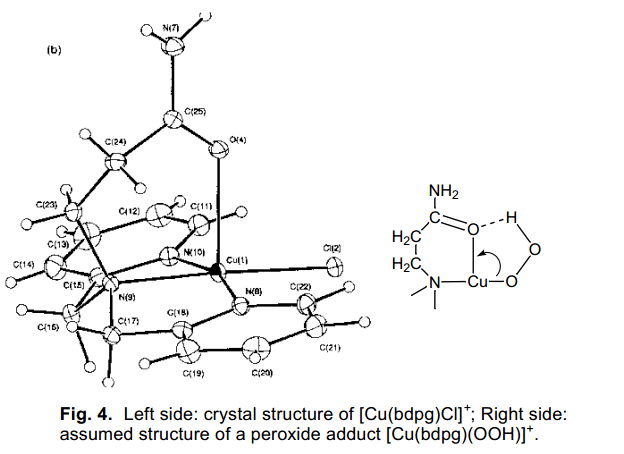
為了得到SOD突變體結構變化和fALS發病機制之間相關的一個全面的解決方案,我們研究了Cu(II)-OOH的反應性能,用來作為一個SOD反應中的重要中間體。為了這個目的,我們已經合成了很多種Cu(II)化合物,這種化合物都含有N,N-雙(2-吡啶甲基)胺配體,如圖3所示。15)所有的Cu(II)化合物結構特點都十分相似(作為例子, [Cu(bdpg)Cl]+晶體結構如圖4所示)。在過氧化氫的參與下,預期生成了加成產物如圖4右邊所示,這個結果與用環己烷等反應一致,并且我們發現Cu(II)化合物過氧化物加合物的反應性能高度依賴于配體系統中的R,這是因為系統中不同的導致Cu(II)離子周圍細微的結構變化的結果。
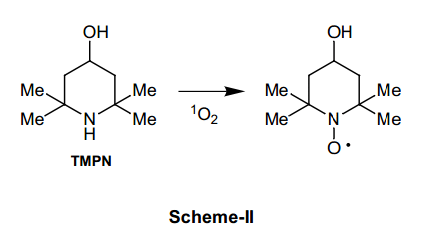
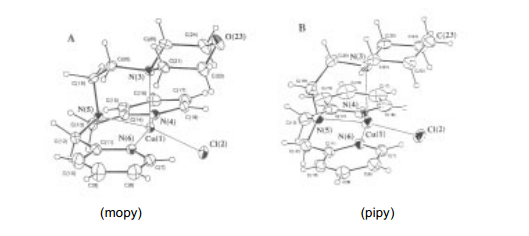
我們已經測定了含有Cu(II)絡合物和自旋捕捉試劑溶液的ESR波譜,例如PBN(α-苯基-N-叔丁基氮氧化物)和TMPN (N,N,N',N'-四甲基-4-哌啶醇), 分別用于OH自由基和單重氮(1?g)(圖表-II)的特定試劑。16)當Cu(II) (tpa) (=三(2-吡啶甲基)胺)或者(bdpg)絡合物與H2O2和PBN混合時,由于PBN自由基的生成,我們檢測不到ESR信號。然而,溶液中含有Cu(tpa)絡合物的情況下,由于TMPN (圖表-II )相應的硝酸自由基的生成,我們能夠檢測到強峰,含有Cu(bdpg)絡合物的溶液則沒有這種情況。尤其是在做Cu(pipy)Cl+和Cu(mopy)Cl+之間的比較時非常有趣。15)這兩種化合物的結構特點非常相似,除了Cu(mopy)Cl+絡合物morphorin 環上的氧原子被替換成了Cu(pipy)Cl+絡合物上面的-CH2的差別(見下圖)。
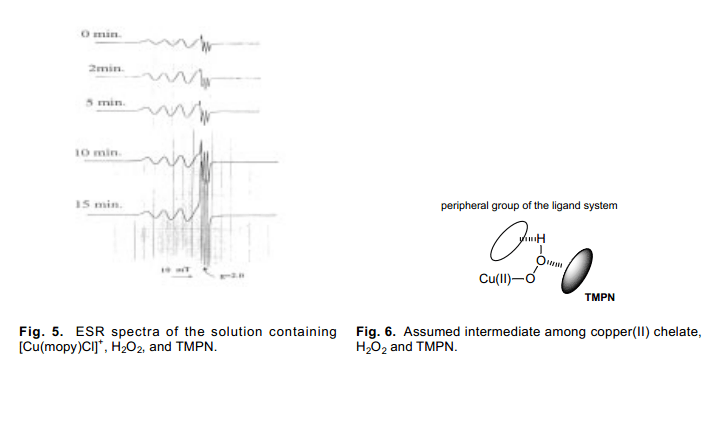
Cu(pipy)Cl+過氧化氫參與條件下, 我們觀測不到硝酮自由基的形成。與此相反,由Cu(mopy)Cl+ 絡合物生成的自由基能夠檢測到有很高的活性,如圖5。在Cu(II)[Cu(Hphpy)Cl]+絡合物里面我們也觀測到了類似的高活性的TMPN 自由基的生成。這種情況下,類似的Cu(mopy)Cl+ 絡合物, 在Cu(II)溶液中加入H2O2,由于銅離子的原因,誘導不了ESR spectrum發生變化;但是添加TMPN 會導致ESR 信號屬性發生急劇的變化( i.e. , 由于銅原子發生超精細結構變化)。上述這些所有的假設是建立在當三種試劑存在于溶液中的時候Cu(II)絡合物、過氧化氫、TMPN的發生(見圖6), 當溶液中中間物形成時可以觀測到過氧化氫獨特的反應性能。
以上事實表明Cu(II)-OOH的反應性能是由中間體的結構性質所決定的(見圖6)i.e. , 通過Cu(II)-OOH,外圍基團和基材之間的相互作用而得。13)在這里應當指出雖然過氧化氫被認為相對惰性且對細胞無毒,但是我們目前的結果明確的表明某些Cu(II)螯合物可以激活過氧化氫,表現出類似于單重氮(1?g)的高活性。
為了進一步獲得Cu(II)-OOH物種反應性能,我們已經測得了Cu(II)化合物和過氧化氫溶液的ESI-質譜。當Cu(Me-bdpg)Cl溶液中加入過氧化氫(見圖3),由ESI-質譜測得生成了[Cu(bdpg)Cl],而不是[Cu(dpal)]。17)這些都清楚的表明Cu(II)-OOH物種能夠切割氧化縮氨酸C-N鍵,而不是水解,因為因為水解斷裂就可能從Cu(Me-bdpg)化合物中得到Cu(dpal)種。
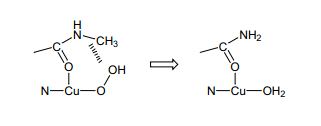
我們還發現某些Cu(II)絡合物在過氧化氫的存在下,蛋白質發生分解,在淀粉樣蛋白beta-肽(1-40)蛋氨酸殘基的氧合硫原子18)處顯示出很高的活性。19)所有的事實可能表明突變型SOD 的“增益功能”是由于在SOD 反應進程中有一個長久的高活性的Cu(II)-OOH作為中間體形成。突變型SODCu(II)周圍的結構發生很大的變化,這給Cu(II)-OOH的反應能行了一個意想不到的影響,正如我們文件所觀測到的。通過Cu(II)-OOH突變型SOD的C-N 鍵的斷裂使得SOD表面發生很大的變化,形成SOD 酶不穩定的二聚體。20)因此,具有高反應性Cu(II)-OOH 基團的形成和存在很有可能是fALS 發病機制中氧化應激的一個內在起源,這與我們最近的含有SOD酶的不穩定性二聚體的研究有一定的關聯。21,22)
3二聚體SOD分子分解成單體
如前文所述,蛋白質聚集被廣泛的認為是很多種神經系統疾病一個常見的病理性特征,包括Huntington’s, Alzheimer’s, and Parkinson’s 等疾病,SOD1聚集體本身的毒性或者是由螯合分子伴侶阻礙蛋白酶體正常的功能運作所產生的運動神經元毒性所致。
2004年, Rakhit 等人報道了SOD1通常是一個二聚體酶,即單體解離之前野生型和突變體蛋白的集合體。23)他們使用“Dynamic Light Scattering (DLS)”方法了檢測二聚體SOD 的單體解離。最近我們也曾報道了毛細管電泳法(CE)是非常適合進行調查研究蛋白質構象變化和蛋白質在溶液中的聚集狀態的一個方法。24)舉一個例子,圖6顯示兩個SOD 和運鐵蛋白的CE曲線圖。25)盡管這兩種酶的濃度時相同的,但是他們的峰的強度彼此有很大的不同,這個已經被合理化的事實是因為溶液中SOD 具有一個剛性二聚體結構,而apo-運鐵蛋白二聚體結構更為靈活。26)

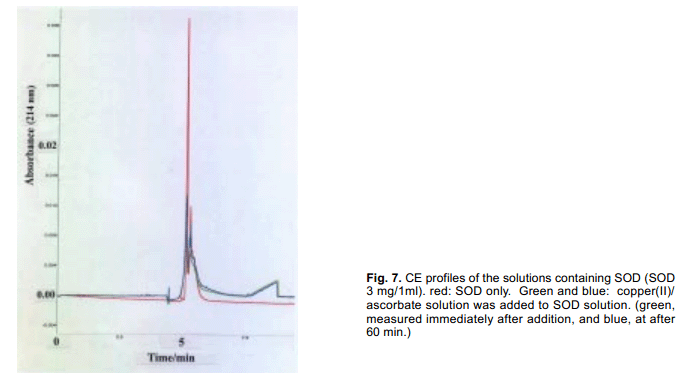
如圖7所示當Cu(II)/抗壞血酸溶液中加入SOD分子的時候,由于二聚體SOD的生成,我們可以觀測到峰值強度急劇下降;我們的實驗系統和Rakhit 等人報道是一樣的。這就清楚的表明二聚體SOD分子的解離可以通過CE法很容易的檢測到。
我們也已經發現過量的過氧化氫導致SOD分子二聚體結構出現松動或者是解離。26)過氧化氫的參與是二聚體SOD 解離的起源是很明確的,26) Kakhit等人很有可能在系統中使用的氧化劑為過氧化氫,偶發性ALS很可能與過氧化氫的存在有關,同樣的討論也可以應用于偶發性朊病毒病的闡述。
通過使用抗體的方法來迅速的純化SOD1,并且用質樸分析法進行耦合連接,Sato等人已經在29 SOD1-突變fALS 患者身上檢測出了 其紅細胞野生型和突變型SOD1相對積累水平。22)他們觀測到檢測不到SOD1突變體的患者持續疾病時間最短。盡管疾病發病年齡與SOD1突變體的數量沒有直接的關系,但是令人信服的是它顯示了疾病時間與突變體積累的一個很強的逆相關系。換句話來說,不太穩定的突變體被發現可能加速病情進展。這一令人驚奇的發現意味著盡管錯誤折疊的SOD1突變體的不穩定形式加劇了疾病潛在毒性的進展,但是SOD1 突變體穩定性與疾病發病沒有一一對應的關系。因此SOD1二聚體分子分裂成為錯誤折疊單體應該會是APS 發病機制過程中必不可少的重要步驟。很明顯過氧化氫在SOD1單體26)形成的過程中起了一個很重要的作用,所以我們應該注意到人體中過量的過氧化氫的生成,特別是二聚體(III)與谷胱甘肽環和其他相關的系統。27)
4.偶發性朊病毒病Cu(II)-OOH
在英國,1980年到大約1996年之間,大約有750,000 牛感染了BSE (牛海綿狀腦病,傳染性海綿狀腦病之一)的牛被屠宰供人類食用,目前人們公認為TSEs 中心部分是將正常的細胞朊蛋白(PrPC)翻譯轉化為一個非正常的的亞型癢病PrP (PrPSc),這種疾病具有一個高-β-折疊片段內容并且與傳染性疾病有關。28)人們普遍的認為PrPC是一種含銅蛋白 (在N-末端的非結構化區域至多有4個銅離子具有八面體重復部位)。小鼠和雞PrPC的重組體的研究分析隨著PrPC銅絡合物的形成使我們得到了一個“增益功能”的重要發現;PrPC 已經被證明有助于直接影響細胞SOD 活性的功能。
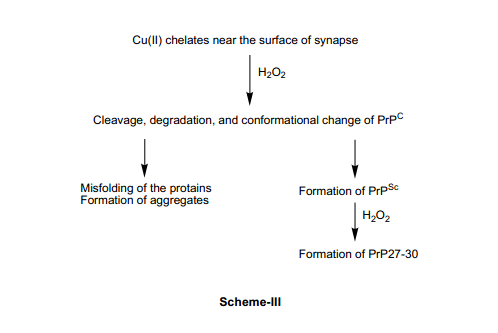
錯誤折疊朊蛋白 (PrPSc) 最終殺死充滿大腦中類似于海綿一樣的神經元和葉狀物。除了PrPSc以外, 還有另外一個從受影響的腦中萃取出來的耐蛋白酶PrP(27-30 kDa), 被叫做PrP27-30。在這里應該注意的是PrP27-30 唯一來自于PrPSc(而不是PrPC), 這個和PrPC至PrPSc直接已經確定順序的氨基酸沒有差別。基于這些事實,我們可以假定,PrPSc銅離子周圍的化學環境應該和PrPC是有所不同的;這種情況和野生型與突變型SOD酶銅離子周圍出現的差異性相似。因此,PrPSc的“增益功能”很有可能歸因于“高活性”Cu(II)-OOH 結構可能發生如描述中的突變體SOD分子,導致在銅離子周圍的肽鍵發生斷裂(大約在90 site處), 然后得到危險的PrP27-30;后面的蛋白質就可能比較類似錯誤折疊SOD單體。除了這一點兒以外,PrPC和PrPSc 中的銅離子很有可能與過氧化氫反應得到Cu(II)-OOH類產物,這很有可能嚴重影響到PrPC 譬如甲硫氨酸殘基的氧合,構象的變化(即PrPSc的形成), 以及過氧化氫參與下蛋白質的降解(見圖表-III)。天然朊蛋白29-32)中觀測到的幾個實驗事實與我們的討論是一致的。所有的這些研究結果都支持著我們的提案,來源于PrPSc SOD功能結構和代謝異常的鐵離子27)的過氧化氫很有可能是零星朊病毒疾病氧化應激力的真正來源。
參考文獻
[1] R. G. Smith and S. H. Appel, Annu. Rev. Med. , 1995, 46,133.
[2] R. H. Brown, Jr., Cell , 1995, 80, 687.
[3] H.-X. Deng, et al., Science, 1993, 261 , 1047.
[4] M. W.-Pazos, et al., Science, 1996, 271 , 515.
[5] I. Fridovich, Annu. Rev. Biochem., 1995, 64, 97.
[6] D. R. Borchelt, et al., Proc. Natl. Acad. Sci. USA., 1994, 91,8292-8296.
[7] L. I. Brujin et al., Science, 1998, 281 , 1851-1854.
[8] J. A. Tainer, E. D. Getzoff, J. S. Richardson, and D. C.Richardson, Nature, 1983, 306 , 284.
[9] A. G. Estevez et al., Science, 1999, 286 , 2498-2500.
[10] M. B. Yim et al., Proc. Natl. Acad. Sci. USA., 1996, 93, 5709-5714.
[11] M. Watanabe et al., Neurobiol. Dis., 2001, 8 , 933-941.
[12] B. Caughey and P. T. Lansbury, Annu. Rev. Neurosci., 2003,26, 267-298.
[13] Y. Nishida and S. Nishino, Z. Naturforsch. , 2001, 56c , 144-153; Y. Nishida, Med. Hypothesis Res., 2004, 1 , 227.
[14] M. B. Yim et al., J. Biol. Chem., 1997, 272 , 8861.
[15] T. Kobayashi, T. Okuno, T. Suzuki, M. Kunita, S. Ohba, and Y. Nishida, Polyhedron, 1998, 17, 1553.
[16] S. Nishino, T. Kobayashi, M. Kunita, S. Ito, and Y. Nishida, Z. Naturforsch., 1999, 54c , 94.
[17] S. Nishino, M. Kunita, Y. Kani, S. Ohba, H. Matsushima, T.Tokii, and Y. Nishida, Inorg. Chem. Communications , 1999,3 , 145.
[18] S. Nishino and Y. Nishida, Inorg. Chem. Communications, 2001, 4, 86; Synth. Reac. Inorg. Metal-Org. Nano-Metal Chem. , 2005, 35, 677.
[19] S. Nishino, A. Kishita, and Y. Nishida, Z. Naturforsch. J. Biosciences, 2001, 56c , 1144.
[20] P. Cioni, A. Pesce, B. Morozzo, S. Castelli, M. Falconi, L. Parrilli, M. Bolognesi, G. Strambini, and A. Desideri, J. Mol.Biol. , 2003, 326 , 1351.
[21] O. Matsumoto and Fridovich, Proc. Natl. Acad. Sci. USA. ,2002, 99, 9010.
[22] K. Yamanaka and D. W. Cleveland, Neurology , 2005, 65,1859.
[23] R. Rakhit et al., J. Biol. Chem., 2004, 279 , 15499-1550.
[24] A. Kishita and Y. Nishida, Annu. Report CIN , 2004, 1; Nishida et al., Synth. Reac. Inorg. Metal-Org. Nano-Metal Chem., 2005, 35, 379; Nishida et al., Z. Naturforsch., 2007, 62b , 205.
[25] Y. Sutoh et al., Chem. Lett., 2005, 34, 141.
[26] Y. Nishida et al., Z. Naturforsch. , 2006, 61c , 273.
[27] Y. Nishida, et al., Z. Naturforsch. , 2007, 62c , 608; Y. Nishida, Recent Res. Devel. Pure & Appl. Chem. , 1999, 3 , 103; Med. Hypothesis Res., 2004, 1 , 227.
[28] B. Caughey, Trends Biochemical Sciences, 2001, 25, 235.
[29] J. R. Requena, N. D. Dimitrova, G. Legname, S. Teijira, S. B. Prusiner, and R. L. Levine, Arch. Biochem. Biophys., 2004, 432 , 188.
[30] H. E. M. MaMahon, A. Mange, N. Nishida, C. Creminon, D. Casanova and S. Lehmann, J. Biol. Chem., 2001, 276 , 2286.
[31] R. Requena, D. Groth, G. Legname, E. R. Sradtman, S. B. Prusiner, and R. L. Revine, Proc. Natl. Acad. Sci. USA., 2001,98, 7170.
[32] N. T. Watt, D. R. Taylor, A. Gillott, D. A. Thomas, W. S. Perera, and N. M. Hooper, J. Biol. Chem. , 2005, 280 , 35914; B. J. Tabler, S. Turnbull, N. J. Fullwood, M. German, and D. Allsop,Biochem. Soc. Trans., 2005, 33, 548; B. J. Tabler, O. M. A.E.-Agnaf, S. Turnbull, M. J. German, K. E. Paleologou, Y. Hayashi, L. J. Kooper, N. J. Fullwood, and D. Allsop, J. Biol. Chem., 2005, 280 , 35789; N. T. Watt and N. M. Hopper,Biochem. Soc. Trans., 2005, 33, 1123; S. Fernaeus, K. Reis,K. Bedecs, and T. Land, Neuroscience Lett., 2005, 389 , 133.
快速導航
化學品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
關于物競
物競數據庫是一個全面、專業、專注,并且免費的中文化學品信息庫,為學生、學者、化學品研究機構、檢測機構、化學品工作者提供專業的化學品平臺進行交流。
數據庫采用全中文化服務,完全突破了中英文在化學物質命名、化學品俗名、學名等方面的差異,所提供的數據全部中文化,更方便國內從事化學、化工、材料、生物、環境等化學相關行業的工作人員查詢使用。
關注我們

-
微信賬號:物競化學品數據庫

-
微博賬號:wjhxp
聯系我們
上海市延長路149號上海大學科技園412室
公司總機: 021-56389801
訂購電話: 4007001514
傳真電話: 021-56389802
客服電話: 021-56332350
電子郵件: wingch@basechem.org
 滬公網安備 31010602001115號
滬公網安備 31010602001115號