以成纖維細胞生長因子為靶標的抑制劑的研究進展
- 2012-08-07
- 專題
哺乳動物成纖維細胞生長因子(fibroblast growth factors,F(xiàn)GFs)家族有18 種成員,它們可以結(jié)合相應(yīng)受體(fibroblast growth factor receptors, FGFRs) 并激活下游信號通路,在胚胎發(fā)生、發(fā)育、血管發(fā)生、血管舒張、神經(jīng)調(diào)節(jié)、缺血保護、創(chuàng)傷愈合和腫瘤發(fā)生等生理和病理過程中起重要作用[1,2]。現(xiàn)已證實,體內(nèi)FGFs/FGFRs 的過度表達與包括腫瘤(如纖維瘤、神經(jīng)膠質(zhì)瘤、黑色素瘤、前列腺瘤、淋巴瘤、白血病、泌尿系統(tǒng)癌等)、骨骼系統(tǒng)疾病(侏儒、顱縫早閉、軟骨發(fā)育不全、黑棘皮癥)和腎衰竭等[1~6]多種疾病密切相關(guān)。以FGFs或FGFRs 為靶標的抑制劑具有治療與FGF/FGFR 過度表達相關(guān)疾病的潛能,其中PI-88、蘇拉明、PNU145156E 、反應(yīng)停等以 FGFs 為靶標的抑制劑已進入臨床試驗[1,6~7]。本文對國內(nèi)外近年來以FGFs為靶標的抑制劑的研究進展進行了綜述。
1 糖類抑制劑
肝素和硫酸肝素(HS)對多數(shù)FGFs 的生物活性是必須的:在肝素/HS 和FGF 的共同作用下,F(xiàn)GFR發(fā)生二聚化,繼而觸發(fā)靶細胞對FGF 的一系列反應(yīng)[1,2,8~10]。肝素和HS都是由兩種相同二糖單元(α-D-氨基葡萄糖(GlcN)(1→4) ?-D-葡萄糖醛酸(GlcA) 和α-D-氨基葡糖(GLcN)(1→4)α-L-艾杜糖醛酸(IdoA)[14])聚合而成的氨基葡聚糖(GAGs)。HS結(jié)合FGF 誘導(dǎo)FGFR二聚化需要糖鏈長度的最低值為八糖,長度小于8 并能結(jié)合 FGF 的肝素寡糖一般表現(xiàn)出對FGF 信號的抑制效應(yīng)[2,11]。有活性的糖類化合物一般都是通過結(jié)合到FGF 的肝素結(jié)合位點而抑制FGF 信號[7,11]。
1.1 抑制FGF的糖類化合物
已報道的糖類抑制劑多含有L-IdoA的硫酸化低聚糖[7,12,13]:二糖結(jié)構(gòu)單元為GlcN(2S,6S)(1→4)IdoA (2S)的六糖能強烈抑制FGF-2和其受體的結(jié)合,抑制人動脈平滑肌細胞增殖(IC50為16μg/mL)[13];二糖單元為α-D- N -乙酰氨基葡萄糖-(1→4)-α-L-IdoA 的非洲蝸牛粘多糖在體內(nèi)能夠明顯抑制FGF-2 刺激的血管增生,抑制鼠黑色素瘤和肺癌的生長[14];4種合成的戊糖(1~4)中,3個糖醛酸殘基全為 L-IdoA 的戊糖1比其它3種戊糖(2~4) 的活性(與FGF-2 的結(jié)合力、抑制 FGF-2和肝素或HS結(jié)合實驗、抑制 FGF-2誘導(dǎo)的人動脈平滑肌細胞的有絲分裂)都要強[12]。不含有L-IdoA的糖及糖衍生物也可以結(jié)合FGF 并抑制其活性,如未硫酸化的單糖噻唑衍生物 5和6能夠明顯抑制 FGF-2 和肝素的結(jié)合,顯著抑制牛動脈內(nèi)皮細胞的生長[15]。在和FGF 的結(jié)合過程中糖殘基種類起到主要作用,但帶負電荷的硫酸根也起到促進和增強它們結(jié)合的作用,甚至一些糖失去硫酸根后對 FGF 無抑制作用[7,11]。過硫酸化的巖藻依聚糖能夠抑制FGF-2誘導(dǎo)的人臍血管內(nèi)皮細胞增殖和血管生成,而脫硫酸的巖藻依聚糖無此活性[16]。Liu 等[11]合成出5種α-D-半乳糖(1→4) ?-D-葡萄吡喃甲苷或者?-D-木吡喃糖甲苷的二糖(7~11),其中化合物7~10和FGF的親和力較強,而R3位無磺酸基的 11 活性遠低于其它二糖。
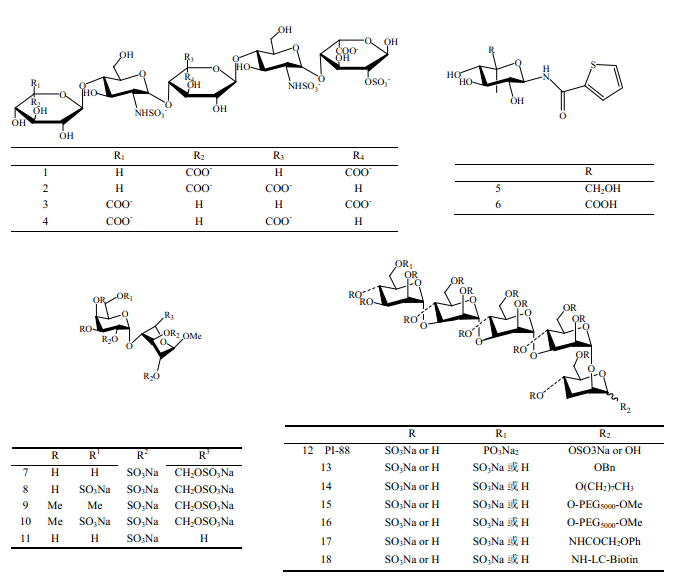
到目前為止,糖類抑制劑方面最有希望的抗腫瘤候選藥物是硫酸化磷酸甘露戊糖 PI-88(12),Ⅰ期臨床試驗表明,它有極好的安全性及耐受性[17~19],在Ⅱ期臨床試驗中,它輔助治療肝癌切除顯示出良好的抗腫瘤復(fù)發(fā)作用[17,19]。PI-88的作用機制主要是通過與 HS競爭結(jié)合血管生成因子VEGF、FGF-1和FGF-2,阻斷生長因子與其受體作用,此外它還可以有效地抑制乙酰肝素酶的活性[17,20]。PI-88對FGF-1和VEGF的結(jié)合力比肝素分別高11倍和3 倍,但是對FGF-2 的結(jié)合力比肝素低13倍[21]。Karoli 等[24,25]以PI-88為先導(dǎo)化合物,設(shè)計合成了19種PI-88衍生物,其中13~18也具有很好的活性,與PI-88相比,它們在拮抗乙酰肝素酶活性方面均保持不變,與VEGF、FGF-1、FGF-2親和力活性方面也基本保持不變,但在藥代動力學(xué)方面改變較大,其中1-O-正辛基衍生物(14)的藥代動力學(xué)有很大提高。
1.2 糖-FGF 相互作用的結(jié)構(gòu)分析
通過對肝素衍生低聚糖-FGF[11]或肝素衍生低聚糖-FGF-FGFR[9,10,23]的X-晶體衍射結(jié)構(gòu)分析發(fā)現(xiàn),肝素衍生低聚糖均是作用于FGF 上帶正電荷的肝素作用位點區(qū)域,因此,糖上的負電荷基團在相互作用過程中起到重要作用。NMR和X 射線晶體衍射分析發(fā)現(xiàn),肝素和 HS中IdoA殘基上的 2-O-磺酸基以及GlcN殘基上的N -磺酸基是和FGF 作用的主要基團,GlcN中的6-O-磺酸基僅與FGF-1相互作用,不與FGF-2 相互作用[11,23]。結(jié)構(gòu)單元為 (4-甲基-2-氨基硫酸根合-6-硫酸根合)-α-D-氨基葡糖(1→4)(2-硫酸根合)-α-L-艾杜糖醛酸甲苷的肝素二糖和FGF-1的分子對接數(shù)據(jù)[11]也證實,糖中磺酸基團和 FGF-1分子中帶正電荷的氨基酸殘基(Lys112、Lys118、Lys113、Lys128、Arg122、Gln127)相互作用。
2蘇拉明類帶負電荷的非糖有機小分子
由于肝素上的硫酸根負電荷在結(jié)合FGF 的過程中起到了重要作用,因此研究者設(shè)計合成了很多帶負電荷的非糖有機小分子抑制劑。
2.1蘇拉明(Suramin)和Suradistas
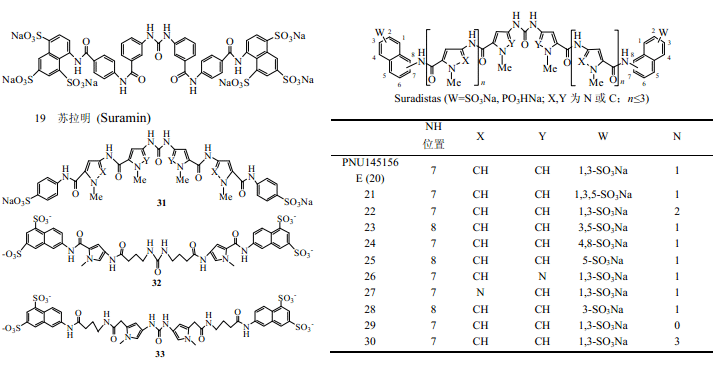
蘇拉明(19)為一種含有萘磺酸基的對稱分子,是一種抑制 FGF等多種與肝素結(jié)合蛋白的生長因子引發(fā)的血管增生的藥物,可以抑制多種腫瘤細胞的增殖,在臨床上用來治療前列腺癌、膀胱癌等多種腫瘤[7,24~30]。但是由于其在體內(nèi)的半衰期長(45~55d),有多個可以作用的靶點,可以抑制一系列重要的酶如DNA聚合酶、蛋白激酶C,和血漿蛋白尤其是白蛋白有高親和力,大劑量使用此藥可以引起凝血紊亂、貧血、腎臟病變,從而表現(xiàn)出比較強的毒性而限制了其臨床應(yīng)用[7,24~30]。
Suradistas(20~33)為蘇拉明的吡咯和吡唑類衍生物[29],Manetti等合成了75個Suradistas類化合物,并對其進行了抑制FGF-2誘導(dǎo)的Balb/c 3T3細胞的有絲分裂和抑制雞胚血管增生等活性篩選。高活性的蘇拉明及Suradistas分子結(jié)構(gòu)特征及其構(gòu)效關(guān)系可歸納為[7,29~34]:(1)帶負電荷的萘磺酸基團可以和FGF中帶正電荷的堿性氨基酸相互作用;(2)分子內(nèi)兩端的萘磺酸基團間間距大小對其活性來說很重要,PNU145156E(20,n=1)活性最好,22(n=2)活性也很好,而29(n=0)、30(n=3)的活性急劇降低,原因可能是分子內(nèi)萘磺酸基團之間的間距只有處于合適長度時,才可以分別與FGF上的肝素結(jié)合位點和受體結(jié)合位點這兩個帶正電荷氨基酸的結(jié)構(gòu)域作用;(3)分子的剛性共平面的共軛結(jié)構(gòu)對其活性來說是必須的,整個分子為非共軛結(jié)構(gòu)的化合物(32、33)的活性降低甚至消失,原因可能是共軛結(jié)構(gòu)中斷的分子末端萘磺酸基團的擺動自由度增加而導(dǎo)致活性降低。
在Suradistas類化合物中20(PNU145156E)以其高效低毒等優(yōu)點而進入了II期臨床研究[29,30]。它能夠高效抑制FGF-2誘導(dǎo)的血管增生、M5076 鼠網(wǎng)狀細胞肉瘤、MXT和S180鼠纖維肉瘤等腫瘤的生長,對M5076/CTX和UV2237纖維肉瘤也有抑制作用;它的毒性小于蘇拉明,雖可誘導(dǎo)血小板短暫的降低但無腎毒性[32,33,36],具有較好的成藥前景。
2.2 其它萘磺酸類化合物
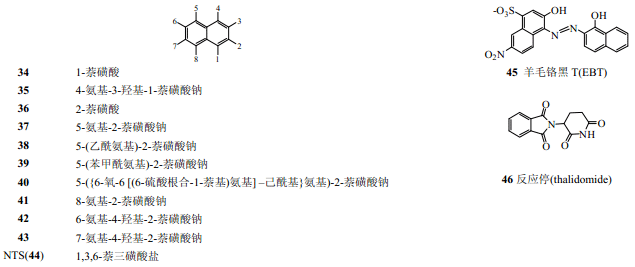
Carlos 等[35]對合成的25個氨基萘磺酸類化合物的活性篩選表明,多個化合物(34~43)都能夠明顯抑制FGF-1誘導(dǎo)的Balb/c 3T3細胞的有絲分裂活性,其中以35的活性最好;37 的活性僅次于 35,而且它在其抑制有絲分裂的濃度范圍內(nèi)沒有表現(xiàn)出細胞毒性[35]。37能夠明顯抑制小鼠局部缺血視網(wǎng)膜疾病中視網(wǎng)膜新生血管的形成[36],在0.008~0.08mg/kg劑量范圍可以抑制皮下埋植含F(xiàn)GF-1或FGF-2的明膠海綿誘導(dǎo)的C57/BI/6鼠新生血管形成,8mg/kg時幾乎全部抑制,而蘇拉明和 NTS(44)要起到相同抑制作用,一般要求其濃度大于 200mg/kg[35]。像蘇拉明一樣,37結(jié)合FGF有可逆性,肝素可以解除其對細胞增殖的抑制活性[38]。NTS 對FGF-1誘導(dǎo)的血管增生和有絲分裂有高效抑制作用[7,37],可以通過提高細胞凋亡抑制兔角膜新生血管生成和神經(jīng)膠質(zhì)瘤生長[38]。羊毛鉻黑 T(EBT)(45)在體內(nèi)也有抑制血管增生和腫瘤生長的作用,在細胞周期的S期阻止人臍靜脈內(nèi)皮細胞增殖,其毒性也低于蘇拉明[7,39]。
2.3 抑制劑-FGF 相互作用結(jié)構(gòu)分析
1H-15N HSQC 核磁共振分析顯示,蘇拉明與hFGF-1的結(jié)合位點分布在肝素結(jié)合區(qū)(Lys127、Tyr139、Lys142)和受體結(jié)合區(qū)(Leu28、Cys30、Gly34、His35) 兩個區(qū)域[31]。但分子對接研究顯示,肝素結(jié)合區(qū)和受體結(jié)合區(qū)的距離大約是3.2nm ,而1個蘇拉明分子長度是2.4nm ,因此不可能1個蘇拉明分子同時作用在2個位點,而是2個蘇拉明分子同時作用在一個蛋白分子表面[31]。用等溫滴定量熱法確證,蘇拉明與hFGF-1按照2?1 的比例以高親和力結(jié)合[31]。分子排阻色譜分析顯示,蘇拉明結(jié)合到 hFGF-1后形成了1個四聚體[31]。通過穩(wěn)態(tài)熒光檢測的熱去折疊實驗分析顯示,蘇拉明誘導(dǎo)FGF 聚合分為2步[31],第1步是2個蘇拉明分子結(jié)合到 1個FGF 上,從而引起了蛋白構(gòu)象的改變,暴露出了蛋白的疏水表面[31],隨后,結(jié)合了蘇拉明的FGF 通過疏水表面自動形成1個無活性的四聚體,不能再結(jié)合 FGFR[31]。
分子對接和分子動力學(xué)實驗[7,30,33,34]顯示PNU145156E 和FGF-1形成1?1的復(fù)合物,通過其萘磺酸基團和FGF-2 上肝素結(jié)合位點(Arg121、Lys126、Asn28) 和受體結(jié)合位點(Arg108) 兩個區(qū)域以氫鍵結(jié)合,另外PNU145156E 上氨基和 Pro133、Gly134通過氫鍵作用。FGF-2 的5 種改構(gòu)體(Cys87羧甲基化,Lys119Gln、Lys125Gln、Lys129Gln、Lys119Gln–Lys129Gln 定點突變 )對PNU145156E 的離解常數(shù)分別提高了12、9.7、9.0、9.1、40.3 倍,而對肝素的離解常數(shù)分別提高了0、10、10、10、和100倍,由此可以說明,PNU145156E 可能通過其分子上的萘磺酸基團結(jié)合于FGF-2 的肝素結(jié)合位點(Lys119、Lys125、Lys129)和Cys87附近的堿性殘基(Arg72、Lys77、Arg81、Lys86)[29]。
化合物37(5-氨基-2-萘磺酸鈉)和FGF-1 的X 射線衍射晶體數(shù)據(jù)分析表明[38],37以1? 1 的比例直接和FGF-1 分子上的肝素結(jié)合位點(Asn32、Lys127、Lys132、Gln141、Lys142)相互作用,前3 個殘基通過氫鍵和磺酸基團作用,后 2 個殘基通過疏水作用和萘環(huán)作用,另外 37 的氨基通過氫鍵和Gly140的羰基作用。NOESY核磁共振分析NTS(44)和FGF-1復(fù)合物三維結(jié)構(gòu)顯示,負電荷的硫酸根和生長因子上正電荷氨基酸Lys127和Lys142相互作用[7,37]。
綜上所述,萘磺酸、NTS等小分子均只結(jié)合到FGF 上的肝素作用位點,而蘇拉明和PNU145156E同時和FGF上的肝素作用位點和受體作用位點發(fā)生作用。蘇拉明和PNU145156E是結(jié)構(gòu)很相似的 2種分子,但蘇拉明-FGF-1和PNU145156E-FGF-2的結(jié)合比例分別為2 ?1和1?1,這種不同可能有多方面的原因,比如FGF 種類的不同、測定條件不同或者模型都有待進一步完善。總之,糖類拮抗劑-FGF 相互作用的結(jié)構(gòu)數(shù)據(jù)不是很成熟,還有待于進一步深入研究。
3 活性短肽
有關(guān)FGF的肽類拮抗劑報道尚不多,其中大部分都是特異作用于受體(FGFR)膜外部分的短肽拮抗劑[41~47]。Fan等[46]應(yīng)用噬菌體篩選技術(shù)篩選到1個活性15肽(PSPVSELSSGRMAYG),與FGFR1的Ⅱ區(qū)180-182和221-223有同源性,能模擬FGFR1上的FGF-1結(jié)合區(qū)域的三維結(jié)構(gòu),與FGF-1上的受體結(jié)合區(qū)域相互作用,抑制FGF-1的促有絲分裂作用。筆者課題組同樣采用噬菌體展示技術(shù),篩選到與bFGF 具有高親和力和最高特異性的小肽P7(PLLQATL),其與FGFR2 D3 256~262這段基序具有較高的序列同源性和相似的高疏水性輪廓,可抑制bFGF 誘導(dǎo)的Balb/c 3T3細胞增殖和雞胚CAM血管生成[47]。
4 其它
反應(yīng)停(thalidomide) (46)是一種II 期臨床用于治療FGF-2 誘導(dǎo)的血管增生抑制劑[6],II 期臨床用于治療前列腺癌[48~51]、腎癌[52,53]、黑色素瘤[54]、小細胞肺癌[55]。能夠顯著降低多發(fā)性骨髓瘤病人血漿FGF-2和VEGF的水平,從而降低病人新生血管的生成,是一種很有希望的治療多發(fā)性骨髓瘤的藥物[56~59]。研究發(fā)現(xiàn),反應(yīng)停能夠抑制不依賴于貼壁的U-87 MG和神經(jīng)膠質(zhì)瘤細胞的生長,而對已經(jīng)敲除FGF-2基因的相應(yīng)的貼壁生長的細胞沒有抑制作用[60]。因此,其治療腫瘤的機制為:反應(yīng)停能夠和FGF-2的啟動子的富G 區(qū)GC boxes (GGGCGG)[60,61]和內(nèi)核糖體進入位點相互作用[61],因而下調(diào)降低 FGF-2的轉(zhuǎn)錄和
翻譯,降低細胞的FGF-2,從而抑制腫瘤細胞的生長和腫瘤新生血管的形成[61]。
5 結(jié)語
FGF 的有機小分子和寡糖類抑制劑多數(shù)是通過電荷和FGF 相互作用,因此在體內(nèi)的靶向性相對較差,毒副作用較大,限制了其應(yīng)用[1,7]。小肽類抑制劑具有用藥劑量小、代謝終產(chǎn)物為氨基酸、毒副作用相對較低等多種優(yōu)點,但小肽類抑制劑在體內(nèi)易受肽酶水解而不穩(wěn)定,從而生物利用度差;小肽的柔性分子結(jié)構(gòu),可具有多種受體分子識別所需的不同構(gòu)象,從而使它們的選擇性降低,導(dǎo)致藥效差,也會產(chǎn)生一些副反應(yīng)。對短肽進行多種化學(xué)修飾后的物質(zhì)為肽衍生物,這類化合物恰能克服普通短肽的以上缺點,同時又可以兼有普通短肽的優(yōu)點,近年來肽衍生物的研究已經(jīng)成為肽類藥物研究的一個熱點,已經(jīng)開發(fā)出的許多肽類藥物都屬于肽衍生物,比如抗腫瘤藥物奧曲肽等。因此,如果把噬菌體展示技術(shù)篩選出來的FGF 拮抗活性小肽改造為肽衍生物,將有可能發(fā)現(xiàn)高效低毒的FGF 抑制劑類候選藥物。由于已報道的FGF 抑制劑基本多是未經(jīng)過分子對接等藥物設(shè)計過程就直接憑經(jīng)驗進行合成和藥理研究,所以將來在進行FGF 抑制劑類藥物設(shè)計時,應(yīng)該充分利用計算機輔助藥物設(shè)計等現(xiàn)代藥物設(shè)計手段,在已經(jīng)發(fā)現(xiàn)的有機小分子抑制劑、寡糖類抑制劑和短肽抑制劑的結(jié)構(gòu)基礎(chǔ)上綜合分析,設(shè)計新的先導(dǎo)化合物。
由于已經(jīng)確證的多種腫瘤、骨骼系統(tǒng)疾病和腎衰竭等都和體內(nèi)多種FGF 過度表達有關(guān)[1~7],因此對FGF 抑制劑的研究將成為治療這些疾病的一個重要方向。然而,到目前為止,F(xiàn)GF 抑制劑的研究主要還是集中在FGF-1 和FGF-2 上,對其它的FGF 的抑制劑的研究不多;盡管已經(jīng)有應(yīng)用于臨床治療研究的FGF 拮抗劑藥物,但是還未發(fā)現(xiàn)很理想的高效低毒拮抗劑藥物。這一切都預(yù)示著對FGF 抑制劑類藥物的研究有著非常廣闊的空間和發(fā)展?jié)摿Α?/p>
參 考 文 獻
[1] A Beenken, M Mohammadi. Nature Rev., 2009, 8(3): 235~53.
[2] M Mohammadi, S K Olsen, O A Ibrahimi. Cytokine, 2005, 16: 107~137.
[3] M Presta, P D Era, S Mitola et al. Cytokine, 2005, 16(2): 159~178.
[4] R Grose, C Dickson. Cytokine, 2005, 16(2): 179~186.
[5] Y H Cao, R H Cao, E M Hedlund. J. Mol. Med., 2008, 86(7): 785~789.
[6] M J Cross, L C Welsh. Trends Pharmacol. Sci., 2001, 22(4): 201~207.
[7] F Manetti, F Corelli, M Botta. Curr. Pharm. Design, 2000, 6(18): 1897~1924.
[8] A D DiGabriele, I Lax, D I Chen et al. Nature, 1998, 393(25): 812~817.
[9] J Schlessinger, Al N Plotnikov, O A Ibrahimi et al. Molecular, Cell, 2000, 6(3): 743~750.
[10] L Pellegrini, D F Burke, F von Delft et al. Nature, 2000, 407(26): 1029~1034.
[11] L G Liu, I Bytheway, T Karoli et al. Bioorg. Med. Chem. Lett., 2008, 18(1): 344~349.
[12] J Kovensky, P Duchaussoy, F Bono et al. Bioorg. Med. Chem., 1999, 7(8): 1567~1580.
[13] Ch Tabeur, J M Mallet, F Bono et al. Bioorg. Med. Chem., 1999, 7(9): 2003~2012.
[14] Y S Lee, H O Yang, K H Shin et al. Eur. J. Pharmacol., 2003, 465(1): 191~198.
[15] A O’ Brien, C Lynch, K M O’ Boyle et al. Carbohydr. Res., 2004, 339(14): 2343~2354.
[16] S Soeda, T Kozako, K Iwata et al. Biochim. Biophys. Acta, 2000, 1497(1):127~134.
[17] V Ferro, K Dredge, L Liu et al. Semin. Thromb. Hemost., 2007, 33(5): 557~68.
[18] L M Khachigian, C R Parish. Cardiovasc. Drug. Rev., 2004, 22(1): 1~6.
[19] R Kudchadkar, R Gonzalez, K D Lewis. Expert. Opin. Investig. Drugs., 2008, 17(11): 1769~76.
[20] L Q Chow, D L Gustafson, C L O'Bryant et al. Cancer Chemother. Pharmacol., 2008, 63(1): 65~74.
[21] S Cochran, C Li, J K Fairweather et al. J. Med. Chem., 2003, 46(21): 4601~4608.
[22 T Karoli, L Liu, J K Fairweather et al. J Med Chem., 2005, 48(26): 8229~36.
[23] M Guerrini, T Agulles, A Bisio et al. Biochem. Biophys. Res. Commun., 2002, 292(1): 222~230.
[24] N Dawson, W D Figg, O W Brawley et al. Clin. Cancer Res., 1998, 4(1): 37~44.
[25] L E Schroder, D Lew, R C Flanigan et al. Urol. Oncol., 2001, 6(4): 145~148.
[26] E M Uchio, W M Linehan, W D Figg et al. J. Urol., 2003, 169(1): 357~360.
[27] X B Zhu, S B Tang, Y Luo et al. Zhonghua Yan Ke Za Zhi , 2005, 41(2): 110~113.
[28] Y Zhang, S Song, F Yang et al. J. Pharmacol. Expt. Thero., 2001, 299(2): 426~33.
[29] M Zamai, C Hariharan, D Pines et al. J. Mol. Struct., 2006, 792-793(3): 23~35.
[30] F Manetti, V Cappello, M Botta et al. Bioorg. Med. Chem., 1998, 6(7): 947~958
[31] K M Kathir, T K Kumar, C Yu. Biochem., 2006, 45(3): 899~906.
[32] K M Kathir, K Ibrahim, T K S Kumar. Biophys. J., 2009, 96(3): 600~601.
[33] M Zamai, V R Caiolfa, D Pines et al. Biophys. J., 1998, 75(2): 672~682.
[34] M Zamai, C Hariharan, D Pines et al. Biophys. J., 2002, 82(5): 2652~2664.
[35] C F Tornero, R M Lozano, M R Horcajo et al. J. Biol. Chem., 2003, 278(24): 21774~21781.
[36] C Lange, Ch Ehlken, G Martin et al. Expt. Eye Res., 2007, 85(3): 323~327.
[37] R M Lozano, M áJiménez, J Santoro et al. J. Mol. Biol., 1998, 281(5): 899~915.
[38] P Cuevas, D Reimers, D Diaz et al. Neurosci. Lett.,1999, 275(2): 149~151.
[39] I Langer, G Atassi, P Robberecht et al. Eur. J. Pharmacol., 2000, 399(2-3):85~90.
[40] H K Fan. Prog. Biochem. Biophys., 2001, 28(3): 338~341.
[41] H Fan, Y Duan, H Zhou et al. IUBMB Life, 2002, 54(2): 67~72.
[42] S Kiyota, L Franzoni, G Nicastro et al. J. Med. Chem,, 2003, 46(12): 2325~2333
[43] I Cosic, A E Drummond, J R Underwood et a1. Mol. Cell Biochem.,1994,130(1) :1~9 .
[44] J Ray,A Baird,H G Fred. Proc. Natl. Acad. Sci. USA,1997,94(13) :7047~7052 .
[45] A Yayon, D Aviezer, M Safran et al. Proc. Natl. Acad. Sci. USA, 1993, 90(22): 10643~10647.
[46] H K Fan, Y Liu, H Zhou et al. IUBMB Life, 2000, 49(6): 545~548.
[47] X P Wu, Q X Yan, Y D Huang et al. J.Cell. Mol. Med., 2008, DOI: 10.1111/j. 1582~4934. 2008. 00506.x.
[48] R J Amato, J H McClain, H Henary. Urol. Oncol., 2009, 27(1): 8~13.
[49] S Romero, G Stanton, J DeFelice et al. Urol. Oncol., 2007, 25(4): 284~290.
[50] M C Cox, W L Dahut, W D Figg. Urol. Oncol., 2006, 24(3): 246~249.
[51] R Dreicer, E A Klein, P Elson et al. Urol. Oncol., 2005, 23(2): 82~86.
[52] T Eisen, C Boshoff, I Mak et al. Brit. J. Cancer., 2000, 82(4): 812~7
[53] P E Clark, M C Hall, A Miller et al. Urology,2004, 63(6): 1061~1065.
[54] A B Reiriz, M F Richter, S Fernandes et al. Melanoma. Res., 2004 , 14(6): 527~31.
[55] R F Riedel, J Crawford, F Dunphy et al. Lung Cancer, 2006, 54(3): 431~432.
[56] C S Mitsiades, P J Hayden, K C Anderson et al. Best Pract. Res. Clin. Haematol., 2007, 20(4): 797~816.
[57] A Jurczyszyn, T Wolska-Smole ń , A B Skotnicki. Przegl Lek, 2003, 60(8): 542~7.
[58] F Bertolini, W Mingrone, A Alietti et al. Ann. Onc., 2001, 12(7): 987~90.
[59] W Du, Y Hattori, A Hashiguchi et al. Pathol. Int., 2004 ,54(5): 285~94.
[60] S C Mei, R T Wu. Mol. Cancer Ther., 2008, 7(8): 2405~14.
[61] T D Stephens, C J Bunde, B J Fillmore. Biochem. Pharmacol., 2000 , 59(12): 1489~99.
注:本文為提供者整理翻譯的,由于知識所限,錯誤在所難免,敬請原諒。如有問題可以查找原文。
統(tǒng)計數(shù)據(jù)
共收錄化學(xué)品數(shù)據(jù)
147579 條
公告
更多>
-
微信掃一掃
關(guān)注:物競化學(xué)品數(shù)據(jù)庫
-
物竟數(shù)據(jù)庫 手機版
國內(nèi)首款化工類專業(yè)手機應(yīng)用
-
微博賬號
wjhxp
快速導(dǎo)航
化學(xué)品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
關(guān)于物競
物競數(shù)據(jù)庫是一個全面、專業(yè)、專注,并且免費的中文化學(xué)品信息庫,為學(xué)生、學(xué)者、化學(xué)品研究機構(gòu)、檢測機構(gòu)、化學(xué)品工作者提供專業(yè)的化學(xué)品平臺進行交流。
數(shù)據(jù)庫采用全中文化服務(wù),完全突破了中英文在化學(xué)物質(zhì)命名、化學(xué)品俗名、學(xué)名等方面的差異,所提供的數(shù)據(jù)全部中文化,更方便國內(nèi)從事化學(xué)、化工、材料、生物、環(huán)境等化學(xué)相關(guān)行業(yè)的工作人員查詢使用。
關(guān)注我們
-
微信賬號:物競化學(xué)品數(shù)據(jù)庫

-
微博賬號:wjhxp
聯(lián)系我們
上海市延長路149號上海大學(xué)科技園412室
公司總機: 021-56389801
訂購電話: 4007001514
傳真電話: 021-56389802
客服電話: 021-56332350
電子郵件: wingch@basechem.org
Copyright ? 物競數(shù)據(jù)庫 2009-2026.All Rights Reserved. 關(guān)于數(shù)據(jù)庫 | 關(guān)于物競 | 法律聲明 | 熱門關(guān)鍵字
 滬公網(wǎng)安備 31010602001115號
滬公網(wǎng)安備 31010602001115號