高效毛細管電泳法在酶反應動力學研究中的應用
- 2012-07-11
- 專題
1967年Hjerten[1]最早提出毛細管電泳技術,并在3mm內徑的管子中使用高場強進行自由溶液的區帶電泳(CZE) 。到了20世紀80年代,毛細管電泳技術取得了突破性的進展,Jorgenson等[2]用75μm 的毛細管,采用柱上熒光檢測成功分離了氨基酸及多肽物質,獲得了40萬的理論塔板數,并實現了正、負離子的同時分離。近幾年來毛細管電泳技術已經成為分析化學中發展最為迅速的領域之一,它具有快速、高分辨率、高靈敏度和樣品用量少、成本低等優點,在生物分析和生命科學領域中有極為廣闊的應用前景。
1 毛細管電泳在酶反應動力學中的應用
酶反應的基本動力學關系,是指酶反應速度與酶和底物間的動力學關系。描述這種基本動力學關系的是米氏方程(Michaelis-Menten equation) 。該方程提供了兩個極為重要的酶反應動力學常數:米氏常數Km 和轉化率Kcat,通過這些參數表達了酶反應性質、反應條件和酶反應速度的關系。其中,Km 是一個特征常數,Km 越大,說明酶和底物的親和力越小,越容易解離。所以,在酶反應動力學中,關鍵是要測得其米氏常數Km。
測定酶反應動力學的方法很多,主要包括分光光度法、熒光法、放射免疫法以及薄層和液相色譜法等。近年來,毛細管電泳法以其進樣量少、操作簡便、易于分離和定量、無需引入有機溶劑等優點,越來越廣泛地應用于酶體系的測定中。毛細管電泳不但具有多種分離模式(如區帶毛細管電泳CZE 、膠束電動毛細管電泳 MEKC 、凝膠毛細管電泳 CGE 、親和毛細管電泳 ACE 、等電聚焦毛細管電泳CIEF、毛細管電色譜 CEC 等),還有多種檢測模式(如紫外、二極管陣列 PDA 、熒光[3]、電化學檢測器等),更加適于酶反應動力學的測定。
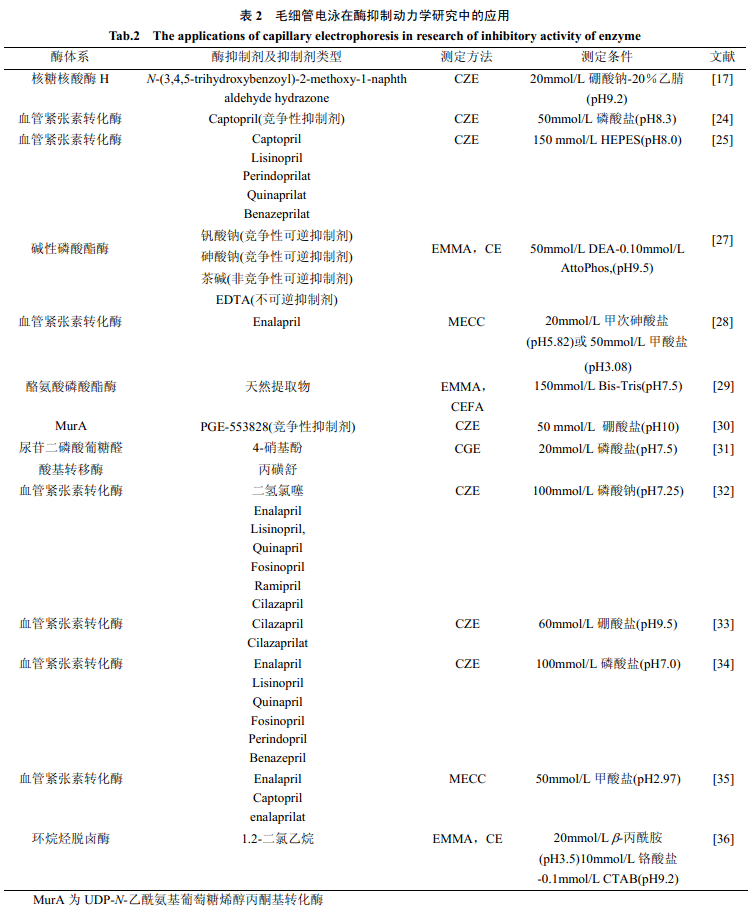
在毛細管電泳法測定酶反應動力學參數的研究中,最常用的測定酶活力的方法是區帶毛細管電泳法[4]。Zhang 等[5]用區帶毛細管電泳法測定了血管緊張素轉化酶(ACE) 的活性、米氏常數和最大反應速度。在式(1)所示酶反應體系中,以產物峰面積對濃度進行回歸分析,得到線性回歸方程,通過監測不同底物濃度下產物的峰面積,計算出酶活力,根據米氏方程用Lineweave-Burk作圖法得到米氏常數和最大反應速度。此法分離重現性高,分析速度快,試劑消耗量少。酶與底物總量僅需要150μL,并且無需加入有機溶劑,6min即可完成分離,8 次測定遷移時間及峰面積的相對標準偏差可分別達到0.15% 和0.71% 。

區帶毛細管電泳法易于控制,但是有些酶反應底物和產物用區帶毛細管電泳法很難達到分離的目的,所以也有不少研究者[6~8]利用膠束電動毛細管色譜法進行分離,被分離組分根據本身疏水性的不同達到分離,疏水性越強,作用力就越大,遷移時間就越長。Choi 等[9]就是采用MEKC分離法,基于膠束和水相的分離,研究測定分泌腺磷酸脂酶的活性。此法能夠準確定量底物和產物,測得值經放射性測定法比較,結果可靠。Yoshimoto等[10]用親和毛細管電泳法進行酶動力學常數的測定,通過橋基基團共價作用將酶鍵合在石英毛細管內壁上,然后引入底物,產物與未反應的底物在電滲流的作用下到達檢測端,由產物的峰高進行定量分析,由Lineweaver-Burk作圖法得到米氏常數,此法節省試劑,僅需要μg級的酶和ng級的底物就可以用來分析測試。
膠束電動毛細管電泳法可以使分子結構相似、分子量相近的底物和產物得到基線分離,但是在酶動力學反應體系中要慎重選用表面活性劑。膠束法最常用的是陰離子表面活性劑SDS ,但SDS 是蛋白質的變性劑,因此選用SDS 測定酶反應動力學參數時,只能適用于柱外反應;有些疏水酶在分離過程中不出峰或者因吸附產生的峰嚴重拖尾,這種情況下可以在緩沖溶液中加入陽離子表面活性劑(如CTAB等)或適量的有機溶劑(如乙腈等),但應注意陽離子表面活性劑極易使電滲反向,檢測時注意加反向電壓;有些非離子表面活性劑(如BRIJ-35) 也可以用于酶的分離與檢測。
在毛細管電泳研究酶反應動力學的過程中,進樣方法是值得探討的問題之一。多數酶反應采用柱外反應(off-line mode),但是柱外反應浪費試劑,所以近年來柱上反應模式(in-line mode) 發展迅速,尤其對于相對不太穩定的物質,用柱上反應法有其特別的優勢[11]。在柱上酶反應體系中,電泳媒介微分析(EMMA) 法[12]是最常用的進樣方法,基于各組分電泳淌度的不同實現柱上反應并得到分離。EMMA法主要有兩種模式:連續模式(continuous mode or long contact mode)和區帶模式(plug-plug mode or transient format or short contact mode) 。連續模式是將一種反應物溶解于背景電解質并填充在毛細管中,另一種反應物以進樣的形式引入到毛細管,兩種反應物在毛細管內充分反應,加電壓實現分離,這種模式比較浪費試劑,所以應用相對較少;區帶模式應用于酶反應主要有三種類型:經典區帶模式(classical plug-plug mode) 、部分填充技術(partial filling technique)和進樣端反應模式(At-inlet reaction)。
有報道[13]采用電泳媒介微分析法(classical plug-plug mode) 與部分填充毛細管技術相結合,可達到反應和分離的目的。電泳媒介微分析法是將底物和酶注入毛細管,淌度較小的先進樣,加一段低電壓,由于電泳淌度不同,兩區帶相遇直至完全重疊,充分反應后,加高電壓實現分離。部分填充技術是指部分毛細管填充酶反應最適緩沖溶液,其它部分填充背景電解質,用于底物和產物的分離。兩種方法結合使得分離快速,易于自動化。
Sigrid 等[14]在研究血管緊張素轉化酶的在線動力學反應中,采用了一種新的三明治式進樣模式(At-inlet reaction),如圖1所示。控制毛細管溫度 37℃,進樣端采用水-酶-底物- 酶-水的夾心模式,等待底物(馬尿酰組氨酰亮氨酸HHL)和血管緊張素轉化酶(ACE) 反應一段時間后,再通入2-[4-(2-羥乙基)-1-哌嗪]乙基磺酸HEPES緩沖溶液進行分離,在230nm 下通過檢測不同底物濃度下產物的峰面積,通過米氏方程,由 Lineweaver-Burk作圖法,得到 1/ v對1/[s]的雙倒數曲線( 其中v為反應速率,[s]為底物濃度),曲線與橫軸的交點即為-1/K m,由此得到米氏常數值。另外,在不影響重現性及分離度的情況下,還可以實現從短端進樣,如在測定PDE 的動力學參數實驗中[15],可以大大縮短分離時間,提高分析速度。
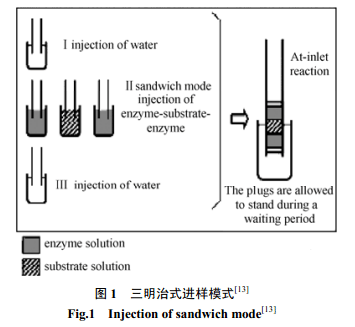
雖然EMMA法較柱外反應法節省試劑,使反應物的用量由μL 變為nL數量級,易控溫,操作簡便,省時,但是由于毛細管內徑較細,柱內反應不夠徹底,也不好控制。夾心式進樣法由于擴散的影響,反應遷移時間的重現性較差;而經典的EMMA法,由于加兩段相差很大的電壓,使得基線不平,檢測靈敏度降低。這些都是需要進一步改進的。
另外,在酶反應動力學的研究中,萬謙等[16]采用多次進樣技術測定蛋白激酶A 活力,發展了連續進樣技術,使能在一個電泳過程中分析十個以上的樣品,通過在線檢測進行積分定量,大大節省了分析時間及費用。
表1 總結了近三年來毛細管電泳技術在酶反應動力學參數測定方面的應用。

2 毛細管電泳在抑制動力學研究中的應用
抑制劑是對酶反應速度有十分重要影響的一種因素,體外測定某些物質對酶反應 K m 的影響,往往有助于推斷該物質可能有的生理效應,同時也可以幫助人們了解酶在機體內可能有的調節功能。抑制是指抑制劑與酶的活性有關部位結合后,改變了酶活性中心的結構(構象) 與性質,從而引起酶活性下降的一種效應。由酶抑制反應動力學可以判斷抑制類型、計算抑制率和半數抑制濃度(IC50)。對于酶反應抑制劑的篩選,可用于研發新藥,對于藥療衛生事業具有很大的意義。
在高效毛細管電泳測定酶抑制劑活性的研究中,一般是基于已經建立的酶反應動力學方法,
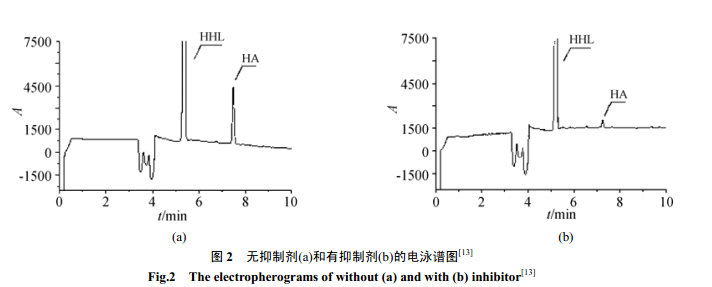
在底物中添加不同濃度或不同類型的抑制劑,同時作對照實驗,根據抑制反應前后峰面積的變化計算抑制率,由抑制率對抑制劑濃度作圖可以得到半數抑制濃度(抑制50%酶活性所需的抑制劑濃度),用來衡量抑制劑對酶的抑制效果,進一步通過抑制反應動力學曲線判斷該反應的抑制類型。辛等[24]基于此法建立了高效毛細管電泳法測定血管緊張素轉化酶抑制劑 Captopril 的活性的方法,經過酶反應動力學判斷Captopril 為競爭性抑制劑,該法簡便快速,7min內即可完成分離,除分析Captopril 對ACE 的抑制活性外,還可分析其它抑制劑對ACE 的抑制活性,并有可能用于篩選新的同類型藥物。Sigrid 等[25]研究了在線抑制動力學檢測法測定ACE 的五種抑制劑,用三明治的進樣模式,柱上反應一段時間后,在230nm下進行分離檢測,結果表明,加入抑制劑后,產物的峰面積比對照組有明顯的減少,如圖 2 所示,抑制效果明顯,由此可通過公式(2)計算抑制率。
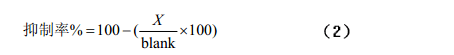
式中X 為加入抑制劑后產物的峰面積;blank為對照組產物的峰面積。
Chang 等[26]建立了用毛細管內微萃取技術測定ACE 抑制劑的活性,復雜樣品無需預處理,4min內即可完成測定,此法快速、選擇性好,適于篩分酶的有效抑制劑。
抑制劑大體可以分為兩種類型——可逆抑制劑和不可逆抑制劑,而根據可逆抑制劑、底物和酶三者的相互關系,可逆抑制劑又可分為競爭性抑制劑、非競爭性抑制劑和反競爭性抑制劑。根據抑制反應動力學及米氏方程可以判斷抑制類型。Angela等[27]研究了毛細管區帶電泳法測定堿性磷酸酯酶的可逆抑制劑、不可逆抑制劑及其活性,采用激光誘導熒光檢測器,酶的抑制作用通過產物所產生的熒光信號的強弱來測定。
表2 是近年來毛細管電泳技術在酶抑制動力學研究方面的一些進展。
3 毛細管電泳(CE)在蛋白質-配體絡合常數測定中的應用
酶與底物,酶與抑制劑的相互作用關系以及相互作用的位點是酶反應動力學必然要探討的問題,所以有必要研究毛細管電泳法在蛋白質-藥物、蛋白質-蛋白質絡合常數測定中的應用。在毛細管電泳法測定蛋白質絡合常數的方法中,主要分為兩類:平衡CE法(equilibrium method)和非平衡CE法(nonequilibrium method)。
3.1 平衡CE法
平衡CE法是將一種絡合組分加入到分離緩沖液中,另一絡合組分以進樣方式進入緩沖液,二者達絡合平衡后進行分離,通過遷移時間的改變或者熒光光譜性質的改變來監測蛋白質絡合的相互作用。平衡CE法主要適用于快速絡合體系,大體有以下三種模式[37,38]:
3.1.1 HD 法(Hummel and Dreyer method) HD 法[39]是將一種物質(蛋白質或配體)加入到緩沖溶液中,另一種物質以少量進樣的方式通過毛細管,兩種物質在毛細管中達到絡合平衡。如果蛋白質-配體絡合物以及自由蛋白質的遷移速度近似相等,并且二者都比未被絡合的配體的遷移速度快,那么蛋白質以及蛋白質-配體絡合物將會和未被絡合的配體相分離,如圖3 所示。由于樣品中總的配體濃度與緩沖溶液中的配體濃度相同,而絡合物的形成和遷移會引起自由配體濃度的改變。因此可以檢測到兩組峰:正峰對應已絡合的配體濃度,負峰對應自由配體濃度。HD法通過分析物峰面積與濃度成線性關系來進行分析測定,可以測得絡合常數。

HD法廣泛應用于可溶性配體和蛋白質絡合常數的測定,尤其對于檢測具有對映體結構的藥物,靈敏度很高。HD法的缺點是自由蛋白和絡合物兩峰難于分離,解決辦法可以通過修飾電荷來改變二者的淌度,或者對蛋白質進行化學衍生,采用熒光檢測,盡可能實現分離。
3.1.2 ACE法(Affinity capillary electrophoresis) ACE 法測定絡合常數的基本原理是基于各物質之間的淌度不同而得到分離的。ACE 法同HD 法相似,也是將一種物質加入到緩沖溶液中,另一種物質以進樣的方式通過毛細管,兩種物質在毛細管中達到絡合平衡。與 HD法不同的是,ACE法是通過未絡合和已絡合分析物的電泳淌度的變化來計算絡合常數。
此法適合研究大陰離子或游離目標蛋白質和配合物間電泳淌度有明顯差別的物質。在蛋白質-DNA ,帶電荷多聚物 -多肽相互作用的研究中應用廣泛。但是 ACE 法不適宜分析高度親和體系,特別是抗原-抗體之間的相互作用[41]。
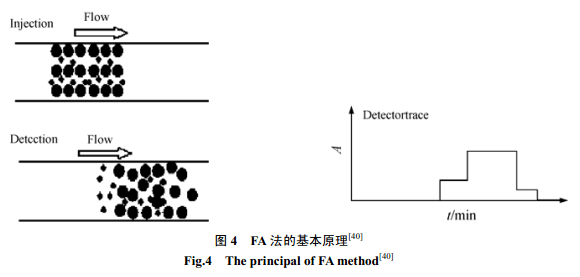
3.1.3 FA 法(Frontal analysis method) FA 法[42~44]是在毛細管中填充緩沖溶液,再將大量樣品(包括相互作用的各個物質)注入到毛細管中。如圖4所示,遷移過程中蛋白質和蛋白質-配體的絡合物由于遷移速度快先到達檢測端,而自由配體分子后到達檢測端,因此檢測端出現兩組峰,通常以自由配體濃度峰的峰高進行定量,可測絡合常數值在103~108范圍內。
FA法的缺點在于進樣量大、試劑消耗多,而且檢出限相對較高。FA法特別適合于高度鍵合的疏水性藥物絡合常數的研究測定。
目前研究比較多的是利用ACE 法測定蛋白質的絡合常數[45~47]。Varenne等[48]用ACE法研究了抗凝血酶與巖藻低聚糖的絡合性質,將不同濃度的多糖注入到背景電解質溶液中,然后采用中性標記物-分離電解質-酶-分離電解質的進樣方式,使酶和電解質中的多糖在毛細管中實現快速的絡合- 解離動力學過程,最終通過有效淌度的計算得到絡合常數。由于蛋白質和配體的快速的絡合- 解離的動力學過程,導致自由蛋白質和其絡合形式的絕對淌度有所差別。蛋白質的峰位置隨著背景電解質中配體濃度的不同而改變,因此,可以通過蛋白質有效淌度的計算求得絡合常數Kf。絡合常數的計算與具體體系相關,計算公式也隨體系的不同而不同。
3.2 非平衡CE法
非平衡CE法是將所有絡合組分預混和,待平衡后,通過電泳使自由組分和絡合組分得到分離。非平衡CE法只能用于解離速率相對較慢的絡合反應,因為解離速率快的反應分離時間過長,導致檢測不到絡合物。隨著 CE分離技術的不斷發展,加上電泳數據分析數學模型的建立,非平衡CE法已經越來越多的應用于動力學研究中[49,50]。非平衡CE法包括NECEEM(nonequilibrium CE of equilibrium mixtures)模式和APCE(affinity probe CE)模式。NECEEM模式靈敏度高,可用于合成核苷親和探針的靈敏度分析[51]。APCE 法理論上可以分離多種蛋白質形式,但是它的條件要求比較苛刻,可行性較差。
Okhonin等[52]用NECEEM法結合SweepCE 法測定了蛋白質-DNA 絡合物二元分子速率常數,使用熒光檢測體系。此法不要求蛋白質和 DNA形成絡合物時熒光光譜發生變化,只要求蛋白質和DNA的淌度不同即可測定。
表3總結了近年來毛細管電泳技術在測定蛋白質-配體絡合常數方面的應用。
4 結語
毛細管電泳法在研究酶反應動力學方面已經取得了相當大的進展,它具有分析速度快、試劑消耗少等優點,但仍然存在一些問題。比如毛細管電泳法最大的缺點就是重現性較差,在酶反應動力學研究中,很多文獻報道的相對標準偏差均大于7%,影響了動力學研究的準確性,是迫切需要改進的。 隨著毛細管電泳技術的不斷發展,一些問題(如靈敏度低,重現性差等)將會逐漸得到解決,該技術在酶反應動力學研究中的應用也將會越來越成熟。特別是多元毛細管電泳,芯片毛細管電泳(MC-CE) 聯用[64]等新技術的提出,更有利于拓展和深入研究各種復雜酶的動力學關系及其應用。
參考文獻
[1] S Hjerten. Chromatogr. Rew., 1967, 9: 122~134.
[2] J W Jorgenson, K D Lukacs. Anal. Chem., 1981, 153: 1298~1302.
[3] M Berezovski, W P Li, P C Dale et al. Electrophoresis, 2002, 23(19): 3398~3403.
[4] J R Anderson, O Cherniavskaya, I Gitlin et al. Anal. Chem., 2002, 74(8): 1870~1878.
[5] R Z Zhang, X H Xu, T B Chen. Anal. Biochem., 2000, 280: 286~290.
[6] V D Sigrid, A V Schepdael, J Hoogmartens. Electrophoresis, 2002, 23: 2854~2859.
[7] M Kulp, M Kaljurand. J. Chromatogr. A, 2004, 1032: 305~312.
[8] M Kulp, M Kaljurand, T Kaambre et al. Electrophoresis, 2004, 25(17): 2996~3002.
[9] S Choi,Y S Lee, D S Na et al. J. Chromatogr. A, 1999, 853: 285~293.
[10] Y Yoshimoto, A Shibukawa, H Sasagawa et al. J. Pharm. & Biomed. Anal., 1995, 13(4/5): 483~488.
[11] N Soň a,V D Sigrid,G Zdenek et al. J. Chromatogr. A, 2004, 1032(1-2): 319~326.
[12] N Soň a, V D Sigrid,V Schepdael et al. J. Chromatogr. A, 2004, 1032(1-2): 173~184.
[13] N Soň a, Z Glatz. Electrophoresis, 2002, 23: 1063~1069.
[14] V D Sigrid, S Vissers, A V Schepdael et al. J. Chromatogr. A, 2003, 986: 303~311.
[15] X Cahours, C Viron, Ph Morin et al. Anal. Chimica Acta, 2001, 441: 15~21.
[16] 萬謙,陳長征,屠紅 等. 生物化學與生物物理學報, 1999 ,27(5):579~584.
[17] K C Chan, S R Budihas, S F J L Grice et al. Anal. Biochem., 2004, 331: 296~302.
[18] E S Dustin, Y Abdelaziez, C H Ahn et al. Anal. Biochem., 2003, 316: 181~191.
[19] Y Kanie, O Kanie. Carbohydrate Research, 2002, 337: 1757~1762.
[20] J Jitkova, C N. Carrigan, C D Poulter et al. Anal. Chimica Acta, 2004, 521: 1~7.
[21] Y Murakami, T Morita, T Kanekiyo et al. Biosen. & Bioelectron., 2001, 16: 1009~1014.
[22] H W Xu, G E Andrew. Anal. Bioanal. Chem., 2004, 378: 1710~1715.
[23] T L Paxon, T S Brown, H N Lin et al. Anal. Chem., 2004, 76: 6921~6927.
[24] 辛志宏,馬海樂,吳守一 等. 藥學學報, 2003, 38(11): 843~843.
[25] V D Sigrid, S Novakova, A V Schepdael et al. J. Chromatogr. A., 2003, 1013: 149~156.
[26] B W Chang, R L C Chen, I J huang et al. Anal. Biochem.,2001, 291: 84~88.
[27] A R Whisnant, S D Gilman. Anal. Biochem. ,2002, 307: 226~234.
[28] E Stellwagen, R Ledger. Anal. Biochem. ,2003, 321: 167~173.
[29] A Belenky, D Hughes, A Korneev et al. J. Chromatogr. A, 2004, 1053: 247~251.
[30] H J Dai, N P Christian, J J Bao. J. Chromatogr. B, 2001, 766: 123~132.
[31] S K Kumiko, K Masaru, T Toyo’oka. J. Chromatogr. A, 2004, 1051: 261~266.
[32] S Hillaert, K D Grauwe,W V Bossche et al. J. Chromatogr. A, 2001, 924: 439~449.
[33] J A Prieto, U Akesolo, R M Jimenez. J. Chromatogr. A, 2001, 916: 279~288.
[34] S Hillaert, W V Bossche. J. Pharm. & Biomed. Anal., 2001, 25: 775~783.
[35] E Stellwagen, R Ledger. Anal. Biochem., 2003, 321: 167~173.
[36] M Telnarova, S Vytiskova, M Monincova et al. Electrophoresis, 2004, 25:1028~1033.
[37] Y Tanaka, S Terabe. J. Chromatogr. B., 2002, 768: 81~92.
[38] M H A Busch, J C Kraak, H Poppe. J. Chromatogr. A, 1997, 777: 329~353.
[39] G Berger, G Girault. J. Chromatogr. B, 2003, 797: 51~61.
[40] R Freitag. J. Chromatogr. B, 1999, 722: 279~301.
[41] R M Seifar, R H Cool, W J Quax et al. Electrophoresis, 2004, 25: 1561~1568.
[42] K L Burns, S W May. J. Chromatogr. B, 2003, 797: 175~190.
[43] T Ohnishi, N A L Mohamed, A Shibukawa et al. J. Pharm & Biomedical Anal., 2002, 27: 607~614.
[44] Y Kuroda, Y Watanabe, A Shibukawa et al. J. Pharm & Biomedical Anal., 2003, 30: 1869~1877.
[45] J Kaddis, C Zurita, J Moran et al. Anal. Biochem., 2004, 327: 252~260.
[46] B Steinbock, P P Vichaikul, O Steinbock. J. Chromatogr. A, 2001,943:139~146.
[47] W B Zhang , L H Zhang , G C Ping et al. J. Chromatogr. B, 2002, 768: 211~214.
[48] A Varenne, P Gareil, S Colliec-Jouault et al. Anal. Biochem., 2003, 315: 152~159.
[49] M BereZovski, S N Krylov. J. Am. Chem. Soc., 2002, 124(46): 13674~13675.
[50] M BereZovski, S N Krylov. Anal. Chem., 2004, 76: 7114~7117.
[51] P L Yang, R J Whelan, E E Jameson et al. Anal. Chem., 2005, 77: 2482~2489.
[52] V Okhonin, M BereZovski, S N Krylov. J. Am. Chem. Soc., 2004,126(23):7166~7167.
[53] M Kinoshita, K Kakehi. J. Chromatogr. B, 2005, 816: 289~295.
[54] M Azad, C Silverio, Y Zhang et al. J. Chromatogr. A, 2004, 1027: 193~202.
[55] G D Li, X J Zhou, Y H Wang et al. J. Chromatogr. A, 2004, 1053: 253~262.
[56] S Kiessig, F Thunecke. J. Chromatogr. A, 2002, 982: 275~283.
[57] K Uegaki, A Taga, Y Akada et al. Anal. Biochem., 2002, 309: 269~278.
[58] C Karakasyan , M Taverna , M C Millot. J. Chromatogr. A, 2004, 1032: 159~164.
[59] Z J Jia, T Ramstad, M Zhong. J. Pharm. & Biomed. Anal., 2002, 30: 405~413.
[60] D W Zhou, F M Li. J. Pharm. & Biomed. Anal., 2004, 35: 879~885.
[61] T Ohnishi, N A L Mohamed , A Shibukawa et al. J. Pharm. & Biomed. Anal., 2002, 27: 607~614.
[62] A Shibukawa , N Ishizawa, T Kimura et al. J. Chromatogr. B, 2002, 768: 177~188.
[63] Y Kuroda, Y Watanabe, A Shibukawa et al. J. Pharm. & Biomed. Anal., 2003, 30: 1869~1877.
[64] A R Stettler, M A Schwarz. J. Chromatogr. A, 2005, 1063: 217~225.
注:本文為提供者整理翻譯的,由于知識所限,錯誤在所難免,敬請原諒。如有問題可以查找原文。
快速導航
化學品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
關于物競
物競數據庫是一個全面、專業、專注,并且免費的中文化學品信息庫,為學生、學者、化學品研究機構、檢測機構、化學品工作者提供專業的化學品平臺進行交流。
數據庫采用全中文化服務,完全突破了中英文在化學物質命名、化學品俗名、學名等方面的差異,所提供的數據全部中文化,更方便國內從事化學、化工、材料、生物、環境等化學相關行業的工作人員查詢使用。
關注我們

-
微信賬號:物競化學品數據庫

-
微博賬號:wjhxp
聯系我們
上海市延長路149號上海大學科技園412室
公司總機: 021-56389801
訂購電話: 4007001514
傳真電話: 021-56389802
客服電話: 021-56332350
電子郵件: wingch@basechem.org
 滬公網安備 31010602001115號
滬公網安備 31010602001115號