“消相反應”在合成中的應用
- 2012-07-09
- 專題
對于比較劇烈的放熱反應,一般通過低溫下緩慢滴加反應物來進行調控。這種反應條件一方面使操作變得復雜,另一方面需要額外消耗能量來維持低溫,這對于大規模工業化生產是非常不利的。近年來,隨著氟化學的發展而誕生的“消相反應”(Phase-Vanishing Reactions)[1]很好地解決了這一問題。利用“消相反應”,可使反應物和底物在反應初期被氟相間隔而不混合,并通過氟相相互進行微量傳遞,進而使得放熱反應在室溫下可控進行。這不僅大大簡化了反應操作,而且無需額外消耗能量。從這點來看,“消相反應”在工業上具有潛在的應用價值。
這一領域的發展始于20世紀90年代初,Vogt 等[2]首先報道了氟溶劑和有機溶劑的相溶性具有溫度依賴性。接著Horvath等[3]發明了氟兩相催化(Fluorous Biphasic Catalysis, FBC) 體系來回收催化劑。這一新概念的提出,立即引起了化學界的廣泛關注。在1996 年后,氟化學在有機合成領域的應用得到了飛速發展,一些重要的進展包括氟相分離(Fluorous Separations) 技術[4~10]、氟相組合化學(Fluorous Combination Chemistry)[11]、氟三相反應(Fluorous Triphasic Reactions)[12]、“消相反應”(Phase-Vanishing Reactions)[1]等。本文主要介紹“消相反應”的研究進展,并簡要介紹筆者在高分子合成方面的初步研究工作,其它方面的研究進展可參見近期的綜述[13~22]。
1 “消相反應”的原理
1.1 基本原理
“消相反應”是氟三相反應的一種變體,其機理有兩種[23]。一是擴散機理,第一相的有機或無機底物擴散到氟相(第二相),然后在第三相與底物反應。在反應過程中,反應物相消失,由此而命名為“消相反應”。另一是萃取機理,先通過溶劑擴散,頂部相密度逐漸增大,底部相密度逐漸減少;當頂部相密度接于中間氟相密度時,就以小液滴的形式穿過氟相到達底物相;小液滴中溶有部分反應物,這樣就使得反應物和底物相接觸并發生反應。擴散機理限于能溶于氟相的底物或反應物,而萃取機理適用于不溶于氟相的反應物。
1.2 基本過程
在“消相反應”中,多使用氟溶劑作為間隔介質,使有機底物和反應物在反應初期不相互混合。這主要是利用氟溶劑特殊的物理性能,即氟溶劑和大多數有機試劑不相溶,而且比大多數有機試劑重[12]。如果反應物(如鹵化物)密度比氟溶劑大,則反應可在試管中進行(圖1)。反應結束后,產物留在上層有機相而底部則出現相消失。如果底物和反應物都比氟溶劑輕,則相消失反應可在U 形管中進行(圖1)。當反應結束時,底物和反應物相中必有一相消失[22]。
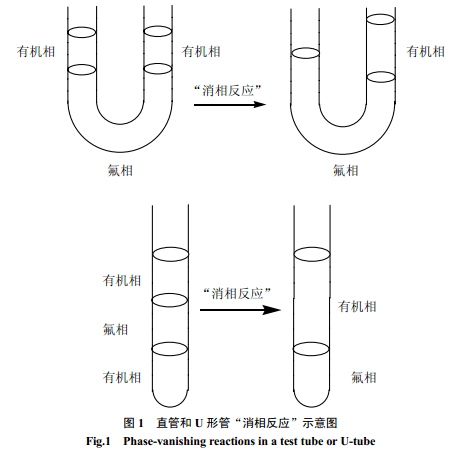
2 “消相反應”的應用
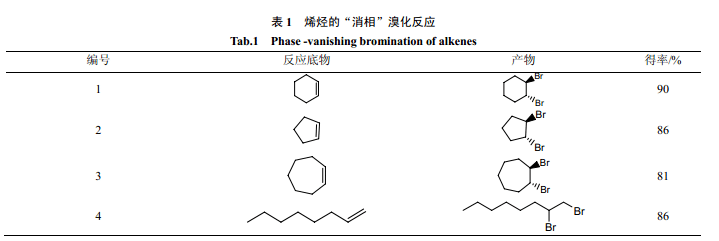
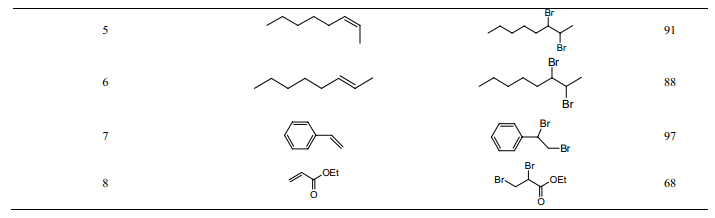
2.1 溴化反應
2002 年,Curran 等[12]最先報道了“消相反應”。他們在室溫下,以全氟己烷為屏蔽相,用液溴對不同的烯烴進行溴化反應(表1)。當反應進行完全時,液溴層消失,氟相恢復透明。研究表明,在保持三相體系條件下,對液溴層進行緩慢的攪拌能大大提高反應速度。
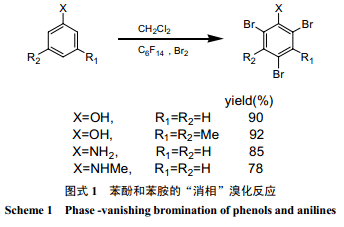
Verkade等[24]利用“消相反應”,用液溴對酚類和苯胺類有機物進行了溴化,得率較高(圖式 1) 。
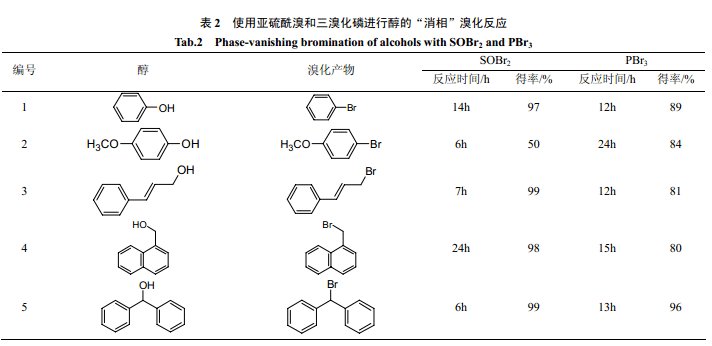
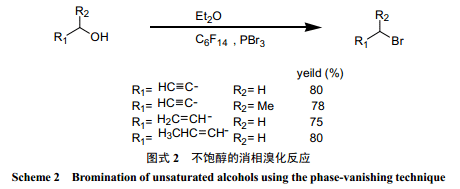
Ryu 等[25]分別用亞硫酰溴和三溴化磷進行了醇的消相溴化反應。在消相反應中,亞硫酰溴的溴化效果明顯好于三溴化磷。另外從反應的得率來看,也比較高。由于消相反應存在傳質過程,所以反應時間要比單一液相反應長。Verkade等[24]用三溴化磷對帶炔丙基和烯丙基的不飽和醇進行了消相溴化反應(圖式 2)。
2.2 氯化反應
Jernej 等[26]在氟三相體系下,用氯氣對烯烴進行氯化反應。這個反應利用了氟溶劑易溶解氯氣的特性。當烯烴被完全消耗后,底物相由于溶解了氯氣而變黃,所以通過有機相顏色的變化可以判斷底物是否反應完全。和許多用氯氣直接氯化的反應相比,在U 形管中通過氟相傳遞的氯化反應有許多優點:第一,反應相和本體反應物是分離的,這點對于像氯氣這樣非常活潑、危險和強腐蝕性的物質來說尤其重要;第二,通過氟相傳遞氯氣的過程是比較緩慢的,這種緩慢加入反應物的過程對于活潑的反應物和放熱反應非常有利;第三,氟溶劑的回收利用率高。

Ryu 等[25]在U 形管裝置中,分別用亞硫酰氯和三氯化磷進行了醇的氯化反應。他們還用亞硫酰氯進行了平行氯化反應,驗證了三相U 型反應體系也適用于平行反應。
2.3 氧化反應
Verkade等[24]用間氯過氧苯甲酸(m-CPBA )進行了胺、硫化物和烯烴的“消相”氧化反應,得率接近文獻報道的最大值。在三相體系中,m-CPBA 溶于密度大于全氟己烷的1,2-二溴乙烷,形成下層有機相;底物溶于一般的有機溶劑,形成上層有機相;氟相作為中間間隔介質。完全反應時,上層或下層有機相消失。后來Curran 指出Verkade進行的“消相”氧化反應機理屬于萃取機理。上層或下層有機相消失取決于最終有機相的密度。Vrkade使用1,2-二溴乙烷來溶解比氟溶劑輕且難溶于氟相的有機物,這一方法大大拓寬了“消相反應”的應用范圍。

2.4 烷基化反應和脫烷基化反應
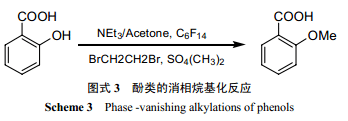
利用硫酸二甲酯進行酚類的烷基化反應一般要在回流條件下進行。但 Verkade等[24]發現,利用“消相反應”,在室溫下就可用硫酸二甲酯進行水楊酸的烷基化反應,得率高于以往文獻報道的最大值(圖式 3) 。
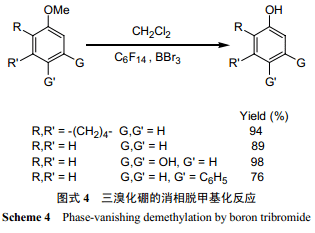
三溴化硼是高效的醚類脫烷基化試劑,而且其密度比全氟己烷大。三溴化硼的脫烷基化反應是個高度放熱反應,一般需要在低溫下進行。但 Curran 等[1]利用“消相反應”,在室溫下用三溴化硼對苯甲醚進行了脫甲基化反應(圖式 4)。
2.5 付氏酰基化反應
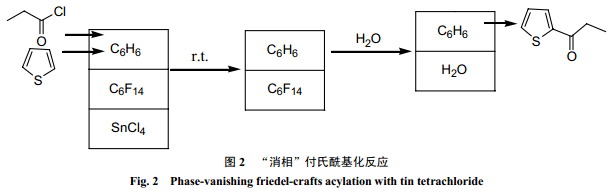
Ryu 等[27]利用消相反應,成功地用四氯化錫對噻吩進行了付氏酰基化反應。在三相體系中,噻吩和丙酰氯溶于苯形成上層有機相,四氯化錫形成底相,全氟己烷作為中間間隔介質。反應結束時,底相消失,產物從上層有機相得到(圖2) 。
3 氟三相體系下甲基丙烯酸甲酯的氧化還原自由基聚合
氟三相體系以及消相反應的應用目前主要集中在有機物合成以及產物分離方面,在高分子合成研究領域的應用尚鮮見報道。筆者在氟三相體系下進行了聚合反應嘗試,并成功地實現了甲基丙烯酸甲酯氧化還原自由基聚合,在引發劑側得到了分子量分布較窄的聚甲基丙烯酸甲酯,多分散系數為1.38 ,反應示意圖如圖 3 所示[28]。

實驗結果表明,氟試劑的屏蔽作用使得引發劑和單體相分離,導致兩側自由基濃度產生巨大差異,并最終使得兩側所得聚合物的分子量和多分散系數明顯不同。引發劑側之所以能得到分子量分布較窄的聚合物,很可能是由于該側自由基濃度大而單體濃度低所致。目前,該聚合體系的動力學正在進一步研究之中,有望產生一種新的高分子合成方法。
4 結語
雖然消相反應體系的研究仍處于初始階段,但是許多研究成果表明消相反應在合成上有著巨大的應用潛力。利用消相反應,在室溫下可使放熱反應緩慢和可控地進行,無需外加設備。消相反應的節能優點,使它具備了大規模工業化生產的潛力。但氟試劑價錢高、環境污染和對人體有害等是該技術的不利因素。反應時間長是該技術的另一個明顯不足。因此,目前該技術的一個重要研究方向是通過調節氟三相體系的溶劑組成、用量和相界面的截面積來提高有機物在氟相的擴散速度,縮短反應時間[23]。相信隨著氟相技術的不斷發展,其工業化進程必將大大加快。
參考文獻
[1] I Ryu, H Matsubara, D P Curran et al. J. Am. Chem. Soc., 2002, 124: 12946~12947.
[2] D W Zhu. Synthesis, 1993: 953~954.
[3] I THorvath, T Rabai. Science, 1994, 266: 72~75.
[4] D P Curran. Synlett, 2001: 1488~1496.
[5] D P Curran, Z Luo. J. Am. Chem. Soc., 1999, 121: 9069~9072.
[6] D P Curran, Y Oderaotoshi. Tetrahedron, 2001, 57: 5243~5253.
[7] W Zhang, Z Luo, D P Curran. J. Am. Chem. Soc., 2002, 124: 10443~10450.
[8] Y Nakamura, S Takeuchi, K Okumura. Tetrahedron Lett., 2003, 44: 6221~6225.
[9] H Matsuzawa, K Mikami. Tetrahed Lett., 2003, 44: 6227~6230.
[10] H Matsuzawa, K Mikami. Synlett., 2002: 1607~1613.
[11] Z Luo, Q Zhang, D PCurran. Science, 2001, 291: 1766~1769.
[12] N Hiroyuki, L Bruno, D P Curran et al. J. Am. Chem. Soc., 2001, 123: 10119~10120.
[13] I T Horvath. Acc. Chem. Res., 1998, 31: 641~650.
[14] E Wolf , G Koten, B Deelman. J. Chem. Soc. Rev., 1999, 28: 37~41.
[15] M Cavazzinni, F Montannari, G Pozzi et al. J. Fluorine Chem., 1999, 94: 183~193.
[16] D P Curran. Med. Res. Rev., 1999, 19: 432~438.
[17] D P Curran. Pure Appl. Chem., 2000, 72:1649~1653.
[18] J A Gladysz, D P Curran. Tetrahedron, 2002, 58: 3823~3825.
[19] C C Tzschucke, C Markert, W Bannwarth et al. Angew. Chem. Int. Ed., 2002, 41: 3964~4000.
[20] A P Dobbs, M R Kimberley. J. Fluorine Chem., 2002, 118: 3~7.
[21] J I Yoshida, K Itami. Chem. Rev. 2002, 102: 3693~3716.
[22] W Zhang. Chem. Rev., 2004, 104: 2531~2556.
[23] D P Curran. Org. Lett., 2004, 6: 1021~1204.
[24] N K Jana, J G Verkade. Org. Lett., 2003, 5: 3787~3790.
[25] H Nakamura, T Usui, I Ryu et al. Org. Lett., 2003 , 5 :1167~1169.
[26] I Jernej, S Stojan, Z Marko. Chem. Commun., 2003, 2496~2497.
[27] H Matsubasa, S Yasuda, I Ryu. Synlett, 2003: 247~249.
[28] S Z Chen, Y P Bai, Z L Li. Chinese Chemical Letters, 2006, 17:258~260.
注:本文為提供者整理翻譯的,由于知識所限,錯誤在所難免,敬請原諒。如有問題可以查找原文。
快速導航
化學品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
關于物競
物競數據庫是一個全面、專業、專注,并且免費的中文化學品信息庫,為學生、學者、化學品研究機構、檢測機構、化學品工作者提供專業的化學品平臺進行交流。
數據庫采用全中文化服務,完全突破了中英文在化學物質命名、化學品俗名、學名等方面的差異,所提供的數據全部中文化,更方便國內從事化學、化工、材料、生物、環境等化學相關行業的工作人員查詢使用。
關注我們

-
微信賬號:物競化學品數據庫

-
微博賬號:wjhxp
聯系我們
上海市延長路149號上海大學科技園412室
公司總機: 021-56389801
訂購電話: 4007001514
傳真電話: 021-56389802
客服電話: 021-56332350
電子郵件: wingch@basechem.org
 滬公網安備 31010602001115號
滬公網安備 31010602001115號