有機合成中的復分解方法的發展
- 2012-06-26
- 專題
1 引言
復分解(Metathesis)這個詞來源于希臘的meta(變化)和thesis(位置) ,是指兩種物質的部分交換。在反應 AB+CD→AC+BD 中,B 已經與C 交換了位置。式1 示出了烯烴交換反應,通過兩個原料烯烴之間的碳烯(亞烷基)交換,形成了兩個新的烯烴。

復分解催化劑已經發展成為有機合成中的極其強有力和多才多藝的工具,它所導致的合成變換令人叫絕。
下面來敘述由肖夫所揭示的復分解機理和隨后由施羅克與格拉布發現的復分解催化劑及其應用。
2 合成方法論的諾貝爾獎
盡管迄今為止,在驚人數量的有機分子中,還只有很少的一部分為合成化學家們所探索,然而,就是這些已經給我們人類提供了許多新的藥物、新的農業化學品和新材料等,如果沒有這些,人類將無法生存。對此的進一步探索必將對人類帶來更巨大的利益,潛力是驚人的。然而,這種進展要求必須要開發新的選擇性合成方法!
自一個世紀前諾貝爾獎建立以來,有機化學的合成方法方面已經獲得了5 次諾貝爾化學獎。所有這些都包括了碳-碳鍵的構建及其化學。對于碳-碳鍵及其在有機合成中的重要性是不言而喻的。1912 年的諾貝爾化學獎授予了 Grignard 和 Sabatier,以表彰他們在確認格氏試劑對于構建分子中碳-碳鍵的形成的重要性以及在不飽和化合物的催化氫化反應中使用金屬所做出的巨大貢獻。1950 年,諾貝爾化學獎授予了 Diels-Alder 反應的發明人,以表彰他們以最簡單的方式重組碳-碳雙鍵而形成新的碳-碳鍵(2個單鍵和一個雙鍵一個)和一個六元環。1979 年,諾貝爾化學獎分別授予了 Brown 和 Wittig 以表彰他們在碳-碳鍵化學(不飽和碳-碳鍵的硼氫化反應及其化學)和其形成(Wittig 反應)方面的貢獻。2001年,碳-碳鍵化學因 Knowles和野依良治(雙鍵還原) 以及Sharpless(雙鍵氧化)對不對稱催化劑方面的工作而再次獲獎。今年,復分解方法(催化打斷碳-碳雙鍵和形成)的發展再次獲此殊榮。
3 烯烴復分解反應的發現
催化復分解反應是隨上世紀 50 年代由齊格勒(Ziegler,1963 年諾貝爾化學獎)在乙烯聚合反應中的觀察而在工業中發現的。盡管在一系列專利中報道了新的過程,但是對其機理并不了解。其中的一個專利是由杜邦公司的 Eleuterio 在 1957 年公布的有關不飽和聚合物的形成。此聚合物是由高張力的原料降冰片烯加到負載在與氫化鋰鋁結合的氧化鋁的氧化鉬上而獲得的[1a]。同一年,申請的另一個有關烯烴歧化反應的專利,即用負載于氧化鋁上的三異丁基鋁和氧化鉬處理使丙烯轉換成乙烯和丁烯,揭示了另一個似乎是新的轉換[1b,c]。
1966 年,那塔(Natta,1963 年諾貝爾化學獎)及其合作者指出,六氯化鎢與三乙基鋁或二乙 基氯化鋁的結合使環庚烯、環辛烯和環癸烯聚合[2]。下一年,Calderon 及其合作者報道了通過采用六氯化鎢和乙基氯化鋁的混合物為引發劑把上述發現延伸到其他的環烯烴[3a,b]。Calderon 認為,環烯烴聚合成多烯單體和脂環烯的歧化是同樣類型的反應,建議稱其為“烯烴復分解反應”[3d]。他們的結果引起了其他科學家對把此反應引入有機和金屬有機反應中的潛力的興趣。然而,對于復分解的機理的理解仍然還是一個謎。
4 復分解是如何進行的-肖夫機理
早期有幾種對烯烴復分解機理的解釋。首先,對于烯烴復分解是否有烷基或亞烷基的交換是有疑問的。Calderon 和 Mol用同位素標記的烯烴的實驗證實,烯烴復分解中相互交換的基團是亞烷基[3a~c]。但是,對于機理中的相互交換以及金屬物種所起的作用起碼還是臆測的。在那時流行的有關機理的幾種說法是用 Calderon 的金屬-配位環丁烯模型[3d]來解釋亞烷基交換的。后來,又提出了Grubbs 的金屬脂環戊烷模型[8a]。這些模型最初往往是不同的作者用不同的方法來描述的(式 2) 。
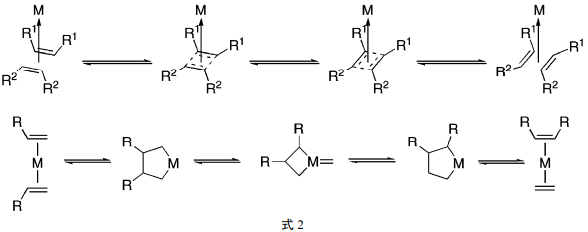
然而,直到法國石油研究所的肖夫對復分解機理的了解的努力以及 Fisher(1973 年諾貝爾化學獎)對于鎢-碳烯復合物的合成、那塔關于通過由WCl6和AlEt3 的混合物催化的開環使環戊烯聚合、Banks 和Bailey 有關用負載在氧化鋁上的W(CO)6催化使丙烯形成為乙烯和2-丁烯的報道之后,才提出了可行的機理。1971年,肖夫和他的學生Jean-Louis Herisson 發表了他們的復分解機理,式3 以修飾的形式示出了他們的機理。

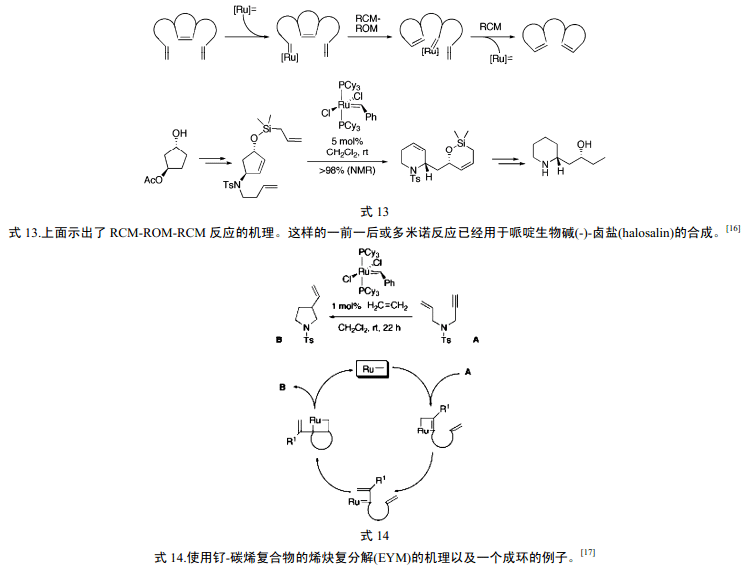
式3 中,金屬碳烯(亞甲基金屬)作為催化劑,把兩個端烯烴復分解為一個內烯烴(E和Z 異構體的混合物)和乙烯。復分解是可逆反應,但是,在這里,乙烯的移走使得反應完全了。
式3b 示出了肖夫催化循環[4] 。亞甲基金屬(金屬碳烯)與烯烴反應,形成了金屬環丁烯中間體。然后,此中間體裂解產生了乙烯和新的金屬碳烯。所形成的乙烯一個來自于催化劑的亞甲基和一個來自于原料烯烴。新的金屬碳烯含有帶有配體的金屬(用[M]表示)和來自于底物烯烴的亞烷基。此亞烷基金屬與一新的底物烯烴分子反應產生另一個金屬環丁烯中間體。在正向反應的分解中,此中間體產生一個內烯烴產物和亞烷基金屬。此亞烷基金屬準備進入另一個催化循環。因此,催化循環中的每一步都包括了亞烷基交換-復分解。
肖夫與其合作者還提出了對此機理的實驗證據,而這些證據不能用其它已經提出的機理來解釋[4]。Grubbs、Katz、Schrock 和其他一些人的實驗也支持了這個機理,因此,現在已經被普遍接受為復分解的機理。
肖夫的亞烷基金屬引發復分解機理告訴我們,人們可以只需合成金屬-亞烷基復合物并讓它們作為催化劑來與烯烴進行復分解反應。[在那時已知的Fisher 型過渡金屬碳烯復合物并不是復分解催化劑。
已經有許多科學家對催化劑的發展做出了貢獻,其中Lappert的有關銠(I)復合物催化劑[5a]、Casey 的鎢復合物[5b]的復分解和其他一些工作是重要的早期貢獻。然而,這里的焦點在于由Schrock 和 Grubbs 在開發對于現代有機合成產生如此巨大影響的金屬-亞烷基復合物方面的突破。
5 Schrock 對于第一個明確、有用的催化劑的創新
盡管許多科學家看好復分解的巨大合成潛力,但是要把它用到有機化學中卻是很復雜的,因為傳統的催化劑對于空氣和濕氣非常敏感而且會引起副反應以及其壽命相對較短。因此,要取得進展就要求要有可以鑒定的、相對穩定的長壽命催化劑,而且甚至其反應性要隨所執行的任務“可調”。
杜邦公司的Schrock 在上世紀70 年代初,試著合成[Ta(CH2CMe3)5],并預期它將是一種穩定的化合物。他確實分離得到的了第一個穩定的金屬-亞烷基復合物,[Ta(CH2CMe3)3(=CHCMe3)],它有高(V)的氧化態[6a]。
然后,Schrock 合成了包括第一個亞烷基復合物在內的其它的鉭-亞烷基復合物,并用 X-線衍射和核磁共振進行了表征。他還發現,形成了金屬環丁烯類。但是,在這些亞烷基復合物中沒有一個可以催化烯烴的復分解[6b]。
1980年,Schrock 與其在麻省理工學院的研究小組報道了一種鉭-亞烷基復合物[Ta(=CHC(CH3)3Cl(PMe3)(O-C(CH3)3)2]催化了順-2-戊烯的復分解。此復合物能夠起作用而其他的鈮和鉭的亞烷基復合物不起作用的原因在于,它存在有烷氧化物配體[6c,d]。
如上所述,在烯烴復分解中,鉬和鎢是活性最大的金屬。Schrock 和他的研究小組加大了他們對尋找穩定分子亞烷基和這些金屬亞烷基復合物的努力,最終得到了以通式[M(=CHMe2Ph)(=N-Ar)(OR)2](其中的R為大體積基團)表示的一系列鉬和鎢的亞烷基復合物。這些復合物是目前已經知道的活性最好的烯烴復分解催化劑(式4) [6e-h]。
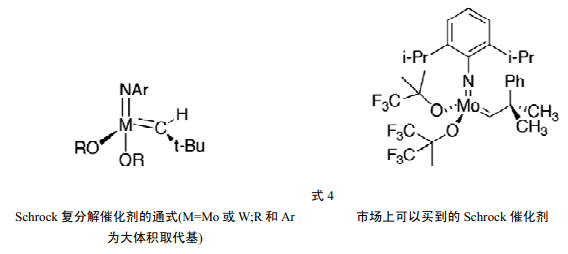
包括斯特拉斯堡的Osborn 和里昂的Basset 也以所報道的鎢復合物對活性烯烴復分解催化劑做出了重要貢獻。
Schrock 催化劑中最有效的是在1990 年報道的,這些催化劑的優點除了它們的活性之外,還在于它們是分子(沒有添加劑)。Schrock 還與Hoveyda 一起開發了不對稱催化復分解的手性催化劑。下面示出了一些應用Schrock催化劑的合成。
6 Grubbs 發現了實用的催化劑
Grubbs 早就對復分解反應感興趣了,比如他就提出過金屬環戊烯中間體的機理[8a]。自在上世紀 80 年代中期開始的對一些用后過渡金屬鹽制備的不太成功的催化劑的探索之后,Grubbs 及其合作者發現,三氯化釕甚至可以在水中使烯烴聚合[8b]。實際上,三氯化釕早已經被Natta用作環丁烯開環聚合的催化劑[7]。Grubbs假設此催化劑也是由金屬碳烯機理來進行的。他們的結果引起了對于可在采用標準有機技術以及對寬范圍官能團適用的催化劑的開發。
作為他們的開發工作的結果,Grubbs 和他的合作者在 1992 年報道了他們的第一個分子釕-碳烯復合物,它不僅對降冰片烯的聚合有活性,而且在存在質子溶劑時也是穩定的[8c]。復合物是亞乙烯基型的[RuCl2(PR3)(=CH-CH=CPh2)],其中的R=Ph(式5)。為了增加催化劑的反應活性,把苯基換成環己基(R=Cy)[8d,e],此變化產生了所期望的反應性、催化了無張力的烯烴聚合和誘導了與脂環烯烴的反應。它促進了許多與Schrock 鉬基亞烷基復合物同樣的反應,而且可以有更大的官能團和可以用標準的有機技術來處置[8f]。Noels 小組在1992 年也報道了環烯烴的Ru-催化開環復分解聚合(ROMP) 。
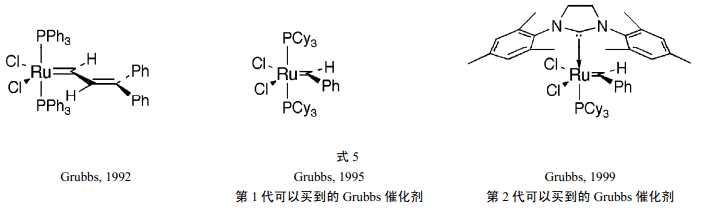
隨著對需要的催化劑數量的增長,要求要有更有效的合成方法。為此,已經開發了一個亞芐基釕復合物的實用途徑。1995 年,Grubbs 報道了新的分子催化劑[Ru(=CHPh)Cl2(PR3)2],R 為苯基或環己基(Cy)[8g,h]。它們的結構與亞烯基的很相近。R 為環己基的化合物已經作為第一代 Grubbs催化劑而商業化(式 5),由于其在空氣中的穩定性和與大量基團的兼容性,此化合物仍是有機化學家們最廣泛使用的復分解催化劑。
在大量的難以關環的反應中,對于在其合理負載量下以高產率得到產物而言,催化劑的壽命仍然是不夠的,需要出現性能改進的催化劑。Grubbs領導的小組通過對機理的深入研究,得到了反應首先包括其一個膦的解離以產生反應性釕中間體的結論。為了加速解離,Grubbs把一個膦用環雙氨基碳烯配體 A 來代替。Herrmann 早已經合成了這種有兩個配體的釕復合物,但是它們的催化劑活性一般。Grubbs的催化劑僅含一個這種配體,加快了余下的一個膦的解離速度,從而加大了其復分解活性。1999 年 Nolan和 Fürstner與 Herrmann幾乎同時發表了類似的結果。新的更高活性的催化劑被稱為第二代Grubbs催化劑。
[RuCl2{C(N(mesityl)CH2)2}(Pcy3)(=CHPh)](式 5)是目前最廣泛使用的交叉復分解反應的有效催化劑[8i] 。此新的釕催化劑具有更好的熱穩定性,現在市場上也可以買到。
Grubbs 的成功也使得其他一些科學家如 Hoveyda、Hofmann、Grela和 Blechert等得到了改進釕基催化劑的靈感和目標。下面示出了一些采用 Grubbs催化劑的合成。
7 應用的多樣性
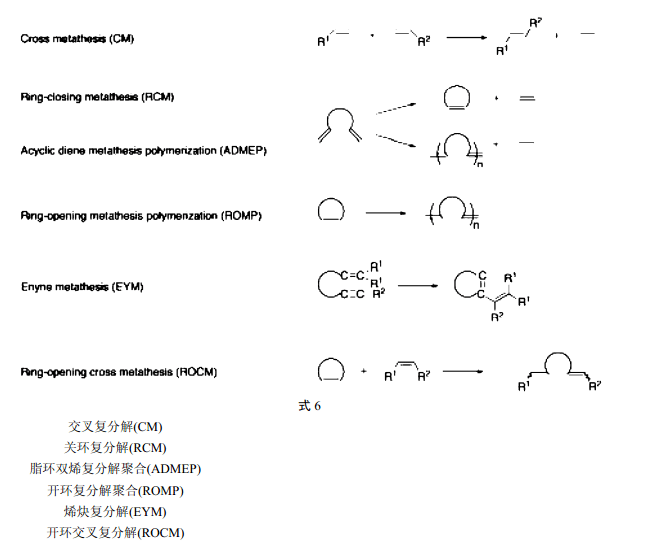
Grubbs 和 Schrock 催化劑給合成化學家提供了新的機會,它們之所以能在有機化學中得到廣泛應用是得益于它們對大量官能團的包容性和效率的結合,而對 Grubbs 催化劑來說,還在于其容易在空氣中處置[9]。式 6 示出了不同類型的復分解反應,隨后的例子說明了新型催化劑的能力。
8 催化劑的進一步發展
催化劑的設計(或在某些情況下的再設計)留給人們巨大的余地,幾乎沒有一個月沒有用于復 分解的新催化劑的揭示。許多這樣的新體系促進和推動了構建高官能化復合分子的努力。復分解所顯現的使轉化成為有效的能力是其它方法所難以達到的,這種趨勢必將持續下去。
9 結果與應用
強調諾貝爾獎得主的發現與改進對于科學研究和商品化合物的工業開發的巨大意義是十分重要的。考慮到 Grubbs 和 Schrock 催化劑的應用時間還很短,所取得的突破是非常驚人的。我們已經目睹了具有特殊性質的聚合物、聚合物添加劑、燃料和生物活性材料比如昆蟲信息素、除草劑和藥物的合成。
由于催化復分解縮短了合成路線,使得反應有更高的產率。這使得我們探索更多更新的有機分子成為可能,從而也可以對“綠色化學”做出貢獻。
參考文獻
[1] (a) Ger. Pat. 1 072 811 (1960) toH.S. Eleuterio, Chem. Abstr., 55 (1961) 16005; U.S. Pat. 3 074 918 (1063) to H.S. Eleuterio.
(b) H.S. Eleuterio, J. Mol. Cat., 65, 55 (1991) and references therein.
(c) R.L. Banks and G.C. Bailey, Ind. Eng. Chem., Prod. Res. Develop., 3, 170 (1964).
[2] G. Natta, G. Dall`Asta, I.W. Bassi, and G. Carella, Makrol. Chem. 91, 87 (1966).
[3] (a) N. Calderon, H.Y. Chen, and K.W. Scott, Tetrahedron Lett., 3327 (1967).
(b) N. Calderon, E.A. Ofstead, J.P. Ward, W.A. Judy , and K.W. Scott, J. Am. Chem. Soc. 90, 4133 (1968).
(c) J.C. Mol, J.A. Moulijn, and C. Boelhouwer, Chem. Commun., 633 (1968).
(d) N. Calderon, Acc. Chem. Res. 5, 127 (1972).
[4] J.-L. Hérisson and Y. Chauvin, Macromol. Chem., 141, 161 (1971). (b) J.-P. Soufflet, D. Commereuc and Y. Chauvin, 276, 169 (1973).
[5] (a) D.J. Cardin, M.J. Doyle and M.F. Lappert, J.Chem. Soc., 927 (1972).
(b) C.P. Casey and Burkhardt, J. Am. Chem. Soc., 96, 7808 (1974).
[6] (a) R.R. Schrock, J. Am. Chem. Soc., 96, 6796 (1974).
(b) C.D. Wood, S.J. McLain and R.R. Schrock, J. Am. Chem. Soc., 101, 3210 (1979).
(c) R.R. Schrock, S.M. Rocklage, J.H. Wengrovius, G. Rupprecht and J. Fellmann, J. Molec. Catal. 8, 73 (1980).
(d) S.M. Rocklage, J.D. Fellman, G.A. Rupprecht, L.W. Messerle and R.R. Schrock, J. Am. Chem. Soc., 103, 1440 (1981).
(e) J.S. Murdzek and R.R. Schrock., Organometallics, 6, 1373 (1987).
(f) R.R. Schrock, S.A. Krouse, K. Knoll, J. Feldman, J.S. Murdzek and D.C. Yang, J. Molec. Catal., 46, 243 (1988).
(g) R.R. Schrock, J.S. Murdzek, G.C. Barzan, J. Robbins, M. DiMare and M. O`Regan, J. Am. Chem. Soc., 112, 3875 (1990).
(h) C.G. Bazan, J.H. Oskam, H.-N. Cho, L.Y. Park and R. R. Schrock, J. Am. Chem.Soc. 113, 6899 (1991).
[7] G. Natta, G. Dall`Asta and L. Porri, Makromol. Chem., 81, 253 (1965).
[8] (a) R.H. Grubbs and T.K Brunck, J. Am. Chem. Soc., 94, 2538 (1972).
(b) B.M. Novak and R.H. Grubbs, J. Am. Chem. Soc., 110, 960 (1988).
(c) S.T. Nguyen, L.K. Johnsson, R.H. Grubbs and J.W. Ziller, J. Am. Chem. Soc., 114, 3974 (1992).
(d) Z. Wu, S.T. Nguyen, R.H. Grubbs and J.W. Ziller, J. Am. Chem. Soc., 117, 5503 (1995).
(e) S.T. Nguyen, R.H. Grubbs,J.W. Ziller, J. Am. Chem., 115, 9858 (1993).
(f) G.C. Fu, S.T. Nguyen, R.H. Grubbs, J. Am . Chem. Soc., 115, 9856 (1993).
(g) P. Schwab, M.B. France and J.W. Ziller, Angew. Chem.. Int. Ed. Engl., 34, 2039 (1995), Angew. Chem. 107, 2179 (1995).
(h) P. Schwab, R.H. Grubbs and J.W. Ziller, J. Am. Chem. Soc. 118, 100 (1996).
(i) M. Scholl, T.M. Trnka, J.P. Morgan and R.H. Grubbs, Tetrahedron Lett.40, 2247 (1999).
[9] Handbook of Metathesis; R.H. Grubbs, Ed.,Wiley-VCH: New York, (2003).
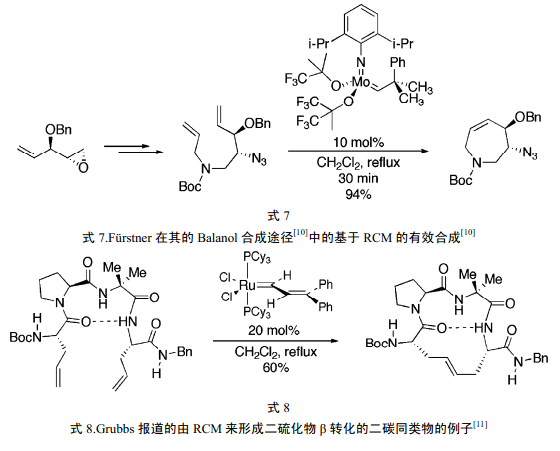
[10] A. Fürstner and O.R. Thiel, J. Org. Chem. 65, 1738 (2000).
[11] S.J. Miller, H.E. Blackwell and R.H. Grubbs, 118, 9606 (1996).
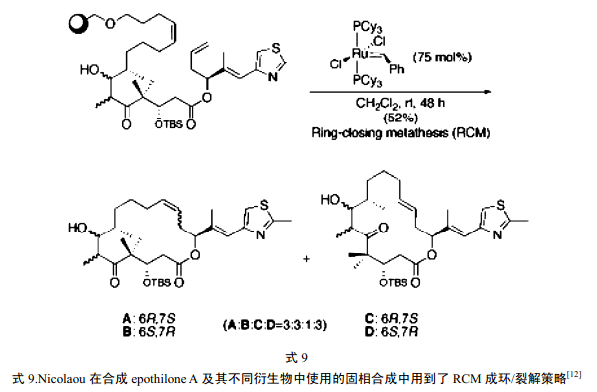
[12] K.C. Nicolaou, N. Winssinger, J. Pastor, S. Ninkovic, F. Sarabia, Y. He, D. Vourloumis, Z. Yang, T. Li, P. Giannakakou and E. Hamel, Nature 387, 268 (1997).
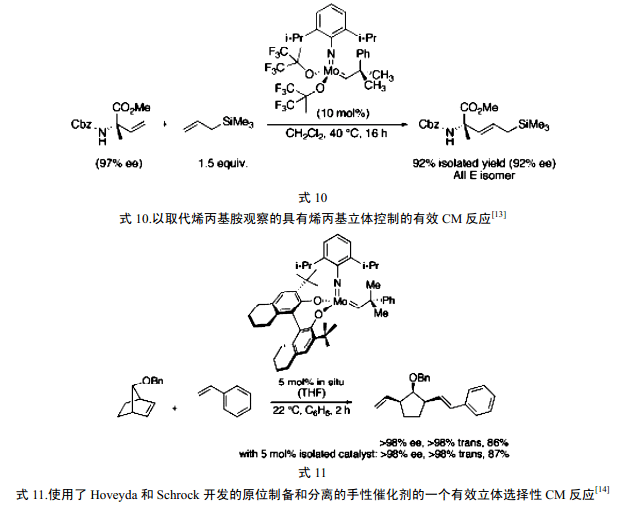
[13] O. Brümmer, A. Rückert and S. Blechert, Chem. Eur. J., 3, 441 (1997).
[14] X. Teng, D. Cefalo, R.R. Schrock and A.H. Hoveyda., J. Am. Chem. Soc., 124, 10079 (2002).
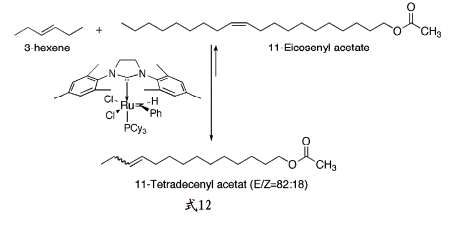
[15] R.L. Pederson, I.M. Fellows, T.A. Ung, H. Ishihara and S.P. Hajela, Advanced Synthesis & Catalysis, 344, 728 (2002).
[16] R. Stragies and S. Blechert, Tetrahedron 55, 8179 (1999).
[17] M. Mori, N. Sakakibara and A.Kinoshita, J.Org.Chem., 63, 6082 (1998).Further reading The Age of the Molecule, N.Hall, Royal Society of Chemistry, London, 1999. Olefin metathesis: Big-Deal Reaction, C&EN, December 23, 2002, p 29-38. Die Olefinmetathese–neue Katalysatoren vergr??ern das Anwendungspotential, M. Schuster and S.Blechert, Chemie in unserer Zeit, 1, 24-29, 2001. Classics in Total Synthesis II, K.C. Nicolaou and S.A. Snyder, Wiley-VCH, Weinheim, 2003.
注:本文為提供者整理翻譯的,由于知識所限,錯誤在所難免,敬請原諒。如有問題可以查找原文。
快速導航
化學品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
關于物競
物競數據庫是一個全面、專業、專注,并且免費的中文化學品信息庫,為學生、學者、化學品研究機構、檢測機構、化學品工作者提供專業的化學品平臺進行交流。
數據庫采用全中文化服務,完全突破了中英文在化學物質命名、化學品俗名、學名等方面的差異,所提供的數據全部中文化,更方便國內從事化學、化工、材料、生物、環境等化學相關行業的工作人員查詢使用。
關注我們

-
微信賬號:物競化學品數據庫

-
微博賬號:wjhxp
聯系我們
上海市延長路149號上海大學科技園412室
公司總機: 021-56389801
訂購電話: 4007001514
傳真電話: 021-56389802
客服電話: 021-56332350
電子郵件: wingch@basechem.org
 滬公網安備 31010602001115號
滬公網安備 31010602001115號