有機修飾及有機物對硅氧烷溶膠-凝膠過程的影響
- 2012-06-11
- 專題
早在 18 世紀中期,人們利用小分子量的四乙氧基硅烷(TEOS)和四甲氧基硅烷(TMOS) 作為前驅體經由溶膠-凝膠過程制備質地均勻的玻璃、陶瓷等材料[1]。目前,經由溶膠-凝膠過程制備的有機-無機復合材料廣泛地應用于光學器件、電子元件、化學傳感、信息存儲、功能涂層和薄膜等領域[2]。為了更好地理解和控制材料的制備條件,人們借助各種表征手段如核磁共振(1H NMR[3]、13C NMR[4]、29Si NMR[5])、FTIR[6]、Raman[7]、SAXRD 和WA X R D[8]、AFM[9]、凝膠滲透色譜[10]、TEM和SEM[11]、光物理手段[12]等對于溶膠-凝膠過程進行大量的機理研究。溶膠-凝膠過程包含水解反應和縮聚反應兩種基本化學反應,硅氧烷水解過程為 SN2 機理[13,14],由此可知取代基的性質及結構必然對水解和縮合反應產生影響;此外,引入的有機組分如能與水解或縮合產物發生相互作用,也必然會影響前驅體的溶膠-凝膠過程。為了較系統地介紹這方面的最新研究動態,本文重點綜述關于有機修飾及有機物對硅氧烷前驅體溶膠-凝膠過程影響以及改性機制的研究。
1 有機修飾硅氧烷前驅體對溶膠-凝膠過程影響
內源性有機修飾硅氧烷是指在小分子硅氧烷前驅體如 TEOS 或TMOS 的基礎上,有機基團取代烷氧基生成不可水解的 Si-C鍵。不同于小分子前驅體,經過有機修飾的硅氧烷因為取代基結構和性質的差異會導致其水解-縮聚反應速度、產物分布、空間結構演化、組織排布方式、產物存在狀態、產物微區環境等方面的差異。
Piana 等[10]利用凝膠滲透色譜跟蹤3-縮水甘油醚丙基三甲氧基硅烷(GPTMS)和甲基丙烯酰氧丙基三甲氧基硅烷(MAPTMS)在相同條件下水解產物分子量的分布,結果發現兩種前驅體水解-縮聚反應產物的物種(單體、低聚物、高聚物)分布各不相同。Fyfe 等[15]利用高分辨率29Si NMR 對甲基三乙氧基硅烷(MTES)和小分子前驅體四乙氧基硅烷(TEOS)混合體系溶膠-凝膠過程的初始階段進行了動力學分析。結果發現,MTES 單體烷氧基每一步水解速度比 TEOS 中烷氧基每一步水解速度快,他們認為甲基的存在對水解產物過渡態具有穩定作用。為了探討有機取代基對硅氧烷溶膠-凝膠過程的影響,Hook[5]對多種有機取代硅氧烷的水解-縮聚速度進行了系統研究,利用誘導效應及立體效應機制解釋取代基對水解-縮聚速度的影響。硅原子的d 軌道和氧原子之間可形成反饋鍵,硅原子上的取代基影響其 d 電子向氧原子反饋,必將影響到氧原子的堿性,即質子化程度。相對乙氧基而言,甲基具有給電子效應,增加硅原子上的電負性,繼而增強氧原子的堿性,提高質子化程度。因此導致甲基取代硅氧烷水解速度較快。相對甲基而言,乙烯基較弱的誘導效應降低了烷氧基的質子化程度,從而使反應速度減慢。由于前驅體水解過程為 SN2 機理,不難理解苯基較大的位阻效應會導致反應前驅體活性降低。
取代基不僅影響水解-縮聚反應的動力學過程,而且這種影響還可導致反應溶膠-凝膠產物的空間結構衍化表現各異。Sugahara 等[16]研究了Si(OEt)4-RSi(OEt)3-EtOH-Water-HCl(R=Me, Ph) 體系在水解-縮聚的起始階段 R 基團對縮合反應生成共聚體的影響。在富水體系中,含有較為疏水的苯基的水解產物在溶液中易于自簇集,而含有甲基前驅體的水解產物易于分散,從而導致甲基三甲氧基硅烷和四甲氧基硅烷水解產物可以形成共聚體,而苯基三甲氧基硅烷和四甲氧基硅烷的水解產物之間不能共聚,只能各自進行縮合反應。說明在利用不同取代基硅氧烷制備共聚雜化材料時,必須依據取代基疏水性選擇水量的多少,以保證不同取代基硅氧烷水解產物的共聚。Mat?jka 等[8]研究了包括高聚物取代基在內的有機取代基對硅氧烷前驅體溶膠-凝膠過程的影響,認為聚合物的生長受控于分子之間縮聚反應和環化的競爭,有機取代基的鏈越長,越容易形成多面體的環形低聚物,主要是T8環或更大的籠狀結構產物。因此較長的取代基增加了低聚物的穩定性,阻礙了縮聚反應,因此體系難以形成凝膠。這與 Voronkov[17]提出的環化反應是縮聚產物衍化成為可溶性多面體晶型低聚物、無定型低聚物或高聚物等不同物理存在狀態的重要因素相符合。
基于有機取代基對水解-縮聚反應和產物結構衍化的影響,不同有機修飾硅氧烷其溶膠-凝膠產物可能具有不同的物理存在狀態。Loy 等[18]在系統研究有機取代基對體系凝膠行為的影響時發現,不同取代基的硅氧烷水解有形成凝膠或者分相形成沉淀兩種趨勢,這與取代基的鏈長、空間位阻有關:含有鏈長較短及位阻較小以基團如甲基、乙烯基、氯甲基的前驅體水解-縮聚反應較快,在產物析出之前已經充分交聯,易于形成凝膠。有趣的是含有如十八烷基、十二烷基長鏈基團的前驅體也容易形成凝膠,他們認為這是長鏈烷基之間的自組織行為使然。而介于兩者之間的取代基則容易導致產物以油或者脂的形式存在。
值得注意的是,疏水性有機修飾的前驅體因為有機基團的存在造成水解產物產生明顯的雙親性,這一特性會導致產物自組織成有序結構。這種作用在含有較長疏水有機取代基修飾的前驅體表現得更為突出。Mat?jka 等[8]在研究中發現,倍半硅氧烷無機骨架和側鏈有機鏈的不相容性導致微區相分離發生,有機側鏈在緊密的網絡微區中自發組織產生膠束結構的有序排布。有機鏈越長,有序性越高,最多的組織排布發生在聚氧化乙烯(PEO)取代的聚硅氧烷中。最近,Hu等[19]借助熒光探針技術在線跟蹤了有機修飾硅氧烷甲基丙烯酰氧丙基三甲氧基硅烷(MAPTMS)在本體相中的水解- 縮聚過程。依據檢測過程中探針熒光發射光譜具有振蕩行為提出,這一前驅體的水解產物具有兩親性質,在本體相中可以依據水量多少而形成油包水型或水包油型膠束結構的簇集體。這說明有機修飾前驅體的溶膠-凝膠過程中有機基團之間存在超分子相互作用是影響產物形態分布的不可忽視的重要原因。
有機修飾對水解-縮聚反應速度和結構衍化的影響必然導致最終產物的性質差異。除了宏觀的表征手段之外,光物理技術是在分子水平上研究材料性質和結構的有力手段[12]。Baker[20]利用芘熒光分子研究了取代基分別為甲基、二甲基、乙基、二乙基、辛基和烯丙基有機修飾硅酸的極性。熒光光譜得到的信息證明了隨著有機基團的鏈長增加,得到的材料的非極性越強。Qian[21]利用溶膠-凝膠過程將熒光物質香豆素440(7- 氨基-4-甲基-1-苯并吡喃-2-酮)包埋于有機修飾硅酸中,研究了引起光譜位移的基質效應和機理。熒光光譜信息表明香豆素440能夠感受到較強的極性,說明體系中含有剩余的硅羥基和水,而在 MTES 和VTES 制備的材料中熒光最大發射波長位移微小,說明材料致密,只含有Si-O-Si網絡結構和甲基或乙基側基。
以上研究都表明有機取代基對前驅體的溶膠-凝膠過程具有較強的影響。目前,設計合成的有機修飾硅氧烷種類很多,對溶膠-凝膠過程的修飾行為各不相同。以下簡單介紹幾類具有典型結構特點的有機取代前驅體對溶膠-凝膠過程修飾行為。
1.1 橋聯型前驅體
橋聯型硅氧烷作為前驅體經由溶膠-凝膠過程制備的橋聯聚倍半硅氧烷是有機-無機雜料中具有特殊性能的一類新型雜化材料[22]。橋聯硅氧烷單體有機連接臂可以是柔性烷基鏈,也可以是不飽和結構的剛性鏈。橋聯硅氧烷在較低濃度水解時即可導致枝化的網絡快速生長,形成凝膠。有機連接臂的長度、剛性、取代基位置等因素可從分子水平上影響材料的結構[23]。連接臂是較短的烷基鏈,得到的聚合物具有多孔結構;連接臂較長,則柔性增加,孔道結構容易塌陷;剛性的共軛結構則會導致材料多孔性。
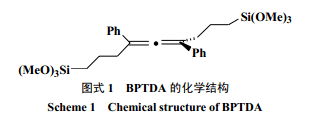
橋聯型硅氧烷前驅體中含有特殊的有機連接臂會為水解縮聚反應過程分子自組裝行為提供可能。Muramatsu[24]利用含有聯苯基連接臂的橋型前驅體[1,4-二(5-三甲氧基硅烷基-1-戊氧基)二苯],經由固態反應制得了具有高度有序性材料。固體反應避免了溶膠的形成,從而削弱了溶劑對材料無規則形成的驅動作用。前驅體本身的晶型結構成為自組織行為的重要因素。固體中分子的結晶形態表面上不能在反應過程中保存,但是可以作為模板或者支架使聚合產物定向生長。取代基為聯苯基、炔基苯基等剛性連接臂都具有上述類似自組織的特性。Cerveau[25]等發現一種具有旋擰線性結構硅氧烷橋聯前驅體[1,3-二(苯基三甲氧基硅烷基)-1,3-二苯丙二烯,BPTDA,圖式[1],其溶膠-凝膠過程具有納米尺度上的自組織行為。前驅體含有扭曲的核心和柔性的側鏈擴展了使體系能夠有序排布的剛性連接臂范疇。有機連接臂之間通過范德華力、π-π堆積等弱的相互作用力誘導了材料結構的有序性使產物具有各向異性。
1.2 含疏水長鏈的前驅體
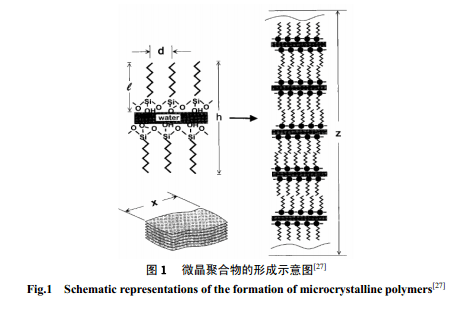
長鏈烷基取代硅氧烷類似表面活性劑的兩親性質和界面行為引起了人們極大的興趣[26]。Parikh等[27]提出在膜-固體基質界面間有超薄的物理吸附水存在時形成的膜有序度最高。形成機制涉及到長鏈硅氧烷水解產物即[n-CnH2n+2-Si(OH)3]在準二維方向上進行的自組裝。水解初級階段較低的水解產物分子在膜-水界面進行類似表面活性劑形成 Langmiur 膜一樣地側向移動而相互靠近,形成覆蓋面較小的膜層。隨水解產物增加而形成高覆蓋面積的單分子膜時,膜層中的水解產物經過硅羥基之間交聯形成 Si-O-Si 鍵。除了在界面上形成 Langmiur 膜的二維結構,基于二維單分子自組裝體的周期性模擬,可以設想 n-CnH2n+2-Si(OH)3 在本體相中能夠形成高度有序的三維相結構。Parikh 等[27]通過研究[C18-Si(OH)
3]的有序排列組裝形成層狀微晶高聚物(圖1) 證實了以上的預想。兩親的硅氧烷分子在氣液界面上進行水解、縮聚,因氣液界面間的惰性氣體分子對兩親的硅氧烷分子的作用較弱,在此條件下兩親硅氧烷分子在氣液界面的有序排列是靠分子自然排布而成的,避免了液固界面上的表面基團反應或基質的引入導致溶液中分子的重排。研究這類硅氧烷在氣液界面上的二維溶膠-凝膠過程有助于深入理解前驅體的水解-縮聚反應機制及反應條件的作用原理。Vidon[28]利用π-Α曲線結合Brewster 角顯微鏡研究了十八烷基硅氧烷(C18TMS)在pH為1.4~12.8 范圍內在水/氣界面上形成單分子膜的過程,結果表明,前驅體無論是在酸性底相還是堿性底相中水解縮合形成的高聚物都為線性結構,歸因于十八烷基相互間的位阻作用使縮聚產物定向生成,疏水基團有序分布在縮聚鏈的兩側。Britt 等[29]研究了十八烷基三甲氧基硅烷(OTMS)和十八烷基二甲基甲氧基硅烷(OMMS)在氣液界面上的質子化、水解和縮合反應的動力學過程。表面壓變化為常數說明硅氧烷中的三個甲氧基是分步進行質子化和水解的。膜層的延展時間、硅烷的堆積密度、底相pH都會影響到二維的水解縮聚過程。
1.3 含有活性基團的前驅體
有機修飾前驅體中含有功能化基團,可以賦予溶膠-凝膠材料獨特的性能。例如,前驅體中含有環氧基可以開環聚合,具有質子傳遞行為[30];氨基的存在為溶膠-凝膠材料鍵合生物分子提供了方便[31];巰基的存在賦予材料具有和重金屬的特異性結合功能[32];含有丙烯基或氨基等活性基團的前驅體可作為偶聯劑增強有機相和無機相之間的復合程度[33],Schmidt 等[34]認為在無機網絡中引入的有機組分主要起到兩個方面的作用:網絡性質修飾或作為網絡結構的一部分。如果有機基團在整個溶膠-凝膠過程中沒有發生化學反應,即“死基”,只是部分阻斷了Si-O-Si 三維網絡結構的形成,可以認為起到了網絡修飾的作用。如果是活性基團則可以參與網絡結構的形成。Mori 等[35]合成了含有多羥基的硅氧烷 N,N-二(2,3-二羥丙基)氨基丙基三乙氧基硅烷,此前驅體在酸性條件下經過水解-縮聚過程形成了平均粒徑為 2.7nm 的顆粒。體系中沒有形成大的團簇是因為烷基鏈功能化基團和硅原子之間通過共價鍵結合,所以顆粒的周圍是高密度羥基功能化烷基鏈,阻礙了粒子之間的相互接觸。同時,Mori 等發現連接在烷基鏈上的羥基可以化學鍵合到水解產物的硅羥基上,驅動了內部環化的產生,得到顆粒內部結構為Si-O-Si 和Si-O-C 組成的含有12~18 個原子的環狀結構。對于含有可聚合功能基的前驅體而言,在溶膠-凝膠過程中通過光、熱、或引發劑引發有機端聚合,因此導致有機網絡和無機網絡同步形成,而且存在著相互競爭[36]。Davis[37]以GLYMO 為前驅體制備有機-無機雜化材料,溶膠-凝膠過程以二乙三胺為催化劑催化環氧基開環聚合。結果證明,無機縮合和有機聚合反應同步進行,在二乙三胺濃度很低的情況下,有機聚合度較低,無機網絡很快形成;高濃度的二乙三胺導致有機網絡的快速形成,但是發現無機網絡的發展緩慢。說明有機網絡和無機網絡的形成存在著相互作用。
1.4 鍵合高聚物的前驅體
高聚物特殊結構特點導致鍵合高聚物的前驅體水解-縮合過程的特殊性。Mat?jka 等[8]通過控制實驗條件,使PEO 鏈節形成晶體,導致水解縮合產物在溶膠-凝膠過程中可能發生自組織行為。他們認為 PEO 鍵合前驅體溶膠-凝膠中間體在熔融態時可以形成球形膠束結構。溫度降低時,分子量為2000 的PEO 鏈段可以完全舒展。而分子量為 5000 的PEO 鏈段可以折疊,形成的聚硅倍半氧烷微區晶體可能是部分互穿平行的結構,或者扭曲的雙層結構。PEO 結晶行為驅動了聚硅倍半氧烷籠型結構排布成為層狀結構。
將嵌段共聚物鍵合到硅氧烷前驅體上,可得到具有新結構特征的雜化材料。Mellon 等[38]通過可逆加成- 裂解鏈轉移(RAFT)方法控制合成了聚甲基丙烯酸甲酯/ 聚甲基丙烯酰氧丙基三甲氧基硅烷[P(MMA-b-MPS)] 嵌段共聚物。P(MMA-b-MPS) 作為溶膠-凝膠過程的前驅體,他們期望通過改變反應條件得到具有可控性構造的嵌段共聚物,使其在溶膠-凝膠材料形成的過程中能夠充分發揮其自組裝的特性,這一研究思想將為結構型納米材料的制備提供一個新途徑。
1.5 其它含有特殊構型的前驅體
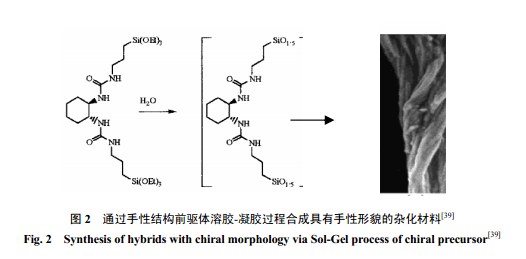
隨著有機-無機雜化材料的廣泛應用,人們別具匠心地設計了很多結構新穎的前驅體。Moreau等[39]設計合成了一種含有手性結構酰胺功能基團的橋型前驅體。他們利用這種前驅體經過溶膠-凝膠過程首次制備了具有手性形貌的雜化材料(圖2) 。就像構建 DNA的雙螺旋結構一樣,氫鍵作用是材料自組織行為的驅動力,在反應過程中手性單體或縮合的低聚物之間的氫鍵弱相互作用可以導向Si-O-Si 鍵的定向生成。這類具有手性形貌和性質的雜化材料在非均相手性催化領域可能有重要的應用價值。Barboiu等[40]利用含有冠醚大環受體的前驅體設計合成了具有超分子結構的有機-無機雜化材料,有機基團之間的氫鍵作用在溶膠-凝膠過程中導致有機凝膠的定向生成。形成的有序性固體膜中的冠醚環對 Na+具有識別作用,基于這種離子識別和傳輸作用可作為三磷酸腺苷的離子驅動泵。這種特殊結構前驅體的研究結果展示了一個功能化有機硅氧烷通過自組裝構建有序結構器件的范例。
2 外源性有機物對溶膠-凝膠過程的影響
除了對前驅體進行內源性修飾之外,外源性有機化合物引入到溶膠-凝膠體系也可以影響溶膠-凝膠過程。外源性有機物引入方式相對于內源性有機基團鍵合到前驅體更為簡單靈活,使外源性有機物對溶膠-凝膠過程的修飾行為也變得異常復雜。下面根據外源性有機物在材料形成過程所起的作用簡要介紹。
2.1 干燥控制添加劑
在溶膠- 凝膠的干燥過程中孔徑變化產生的壓力或溶劑揮發產生的毛細作用使凝膠塊體產生裂紋或碎片,解決這一難題的有效方法之一是在材料的溶膠階段加入干燥控制化學添加劑(DCCA)[41]。如甲酰胺、N,N-二甲基甲酰胺、乙腈、草酸等。DCCA 的作用機理較為復雜,一種看法是,DCCA的加入使材料具有孔徑分布較窄的大孔結構,從而減弱了毛細作用,另一種看法是,DCCA 和硅酸表面上的羥基形成氫鍵可以阻止水分子和 Si-OH作用,且空隙中充滿 DCCA 比充滿水的表面張力小,從而使水分子容易揮發[42]。Viart 等[43]利用測試 pH和FT-IR 研究甲酰胺對 TEOS 溶膠-凝膠過程的作用機制時發現,沒有甲酰胺存在時,前驅體縮合易生成線性產物,交聯度較低;甲酰胺的存在使前驅體易生成小于6個硅原子的低聚物,在這些小的低聚物基礎上進行再縮合增大了凝膠網絡的交聯度,使SiO2 網絡結構趨向玻璃化。Lenza 等[44]在研究某些 DCCA 對溶膠-凝膠膜材料結構的影響時發現,DCCA 的存在下,較高溫度制備的材料表面無裂紋的塊體結構和介觀尺度的大孔隙結構材料,且材料存在大量的自由硅羥基。
2.2 水解-縮聚反應調節劑
某些有機添加物可以對硅氧烷的溶膠- 凝膠速度施加影響。如對含有可聚合的環氧基前驅體(GLYMO)而言,1-甲基咪唑(MI)、3-氨丙基三乙氧基硅烷(APTES)和三氟化硼乙醚絡合物[BF3(OEt)2]對其水解-縮合反應都會產生影響[37]。MI能夠有效催化環氧基開環,但不直接影響網絡結構的形成;APTES中的氨基不僅具有催化溶膠-凝膠過程作用,而且因分子中烷氧基存在,可參與凝膠網絡的形成。BF3(OEt)2 具有有效提高無機端的交聯度和聚氧化乙撐衍生物形成的雙重功能。
值得關注的是,基于和前驅體分子中有機基團之間的相互作用,某些有機物可以對含有較長疏水鏈修飾的前驅體在氣/液界面上的水解-縮聚反應產生影響。Park 等[45]研究了酸性表面活性劑全氟代十四烷酸(PFTDA) 對氣/ 液界面上形成的十八烷基三甲氧基硅烷(ODTMS)單分子膜膠凝行為的影響,結果表明加入2(mol)% PFTDA 即可加速 ODTMS 凝膠形成,他們認為 PFTDA在單分子層中快速擴散,質子很快傳播到反應活性位點甲氧基附近,導致水解反應部位酸度很高,從而催化了ODTMS 的水解。通過Cd2+與PFTDA形成絡合物從而減弱了 PFTDA催化活性的實驗結果證實上述推測。Wang等[46]利用10,12-二炔基-二十五烷酸(PDA)和它的衍生物N-(11-O-α-D-吡喃葡糖-3,6,9-三氧雜)十一烷基-10,12-二炔基-二十五烷酸(PDTMC)以及十八烷基三甲氧基硅烷(ODTMS)構成Langmuir 單分子膜(圖3) ,混合物之間存在兩種作用力:一方面較長碳鏈之間的疏水相互作用;另外ODTMS 單體水解產物硅羥基和 PDA 分子中羥基以及 PDTM 分子中酰氨基之間存在氫鍵作用。因此適當的 pH 條件下,混合物互溶沒有相分離發生時,彼此之間產生了立體位阻效應,即“空間隔離效應”,導致 PDA/PDTM的光聚合反應和 ODTMS 的縮和反應能夠相互影響。利用這一效應來調節硅氧烷溶膠-凝膠反應的有機物還有硬脂酸甲酯[47]等一些惰性有機物。

2.3 模板及結構導向劑
有機物對溶膠-凝膠過程中的水解-縮合產物往往表現出模板或結構導向作用。利用有機物作為模板劑合成各種孔道結構的溶膠-凝膠材料的相關工作已經有大量的文獻報道[48-50]。模板可以是以共價鍵維系特異形貌的具有不同空間結構的硬模板,如高分子聚合物[51]、水凝膠[52]或有機凝膠[53];也可以是通過分子間或分子內的弱的相互作用力而形成的分子自組裝體或超分子模板[54-55]。這里,筆者著重介紹有關在溶膠-凝膠過程中有機物發揮模板作用或結構導向劑作用的機理研究。Beck等[50]以表面活性劑形成的液晶為模板合成了一系列 MCM-41 型介孔分子篩,并且提出了表面活性劑簇集形成六角形的棒狀膠束導致材料特殊形貌形成的機理。實驗證明,材料形成的結構和孔徑與表面活性劑的鏈長及溶解性密切相關。Sahu 等[56]利用香豆素 480 跟蹤了表面活性劑CTAB 存在下,溶膠-凝膠過程中溶劑化動力學過程時發現,CTAB 的極性頭基和硅羥基之間的相互作用使包埋在其中的水運動受限,探針分子的各向異性值充分證明了疏水核心的存在。最近,Zhang 等[57]以一種類似“蜥蜴”型的分子作為模板設計合成了功能化的介孔二氧化硅材料。模板分子為兩親物,陽離子頭基共價結合于可縮合的硅氧烷基團;中間為欲賦予材料的功能基團;尾部為疏水性的烷基鏈,且在分子固定后可在酸性條件下水解實現“剪切”。在“剪切”前的疏水尾可以保證前驅體具有自組裝性質,在固定后剪切使功能基釋放裸露于材料孔道中。該方法為具有特殊分子結構新型模板的設計開辟了一條新途徑。Chen等[58]以非離子三元嵌斷共聚物聚氧化乙烯-聚氧化丙稀-聚氧化乙烯(EO20 PO70 EO20)和陰離子表面活性劑為結構導向劑經由 TEOS 溶膠-凝膠過程制備具有雙連續立方結構的介孔二氧化硅材料,結果發現,調節共聚物和表面活性劑的含量可以提高溶膠-凝膠材料的有序性,說明是二者共同組織的膠束簇集體發揮了模板的作用。Shinkai等[59]]首次利用不含氨基和靜電荷的有機分子和苯胺類有機溶劑形成的有機凝膠為結構導向劑經由 TEOS 溶膠-凝膠過程合成了纖維狀二氧化硅材料。不同于以往的氫鍵作用,苯胺分子和膠凝劑分子之間的疏水相互作用以及 π-π 弱相互作用是模板結構轉錄到二氧化硅材料中的驅動力。
類似于生物的礦化作用,在溶膠-凝膠過程中有機物和無機物能夠實現在分子水平上的穿插復合本身就意味著相互間的導向作用。孫繼紅等[60]研究了聚氧乙烯(PEO)-聚氧丙醚(PPO)-聚氧乙烯醚(PEO)(簡稱 EPE)三元嵌段共聚物作為改性劑對溶膠-凝膠過程的修飾行為。EPE 首先與體系中微量水相互作用,通過影響 TEOS 的水解縮聚反應,改變了Si 的配位環境,EPE 疏水基和親水基的存在使得基本結構單元形成彎曲度較大的鏈狀結構,從而導致SiO2 溶膠的網絡結構。Shchipunov 等[61]利用各種天然多糖大分子和可溶性前驅體(TEOS 的低聚物)合成了新一類塊體硅酸生物材料,認為大分子結構中的羥基和硅醇之間的氫鍵作用或形成共價鍵為二氧化硅顆粒的生長提供了“晶核”,而且在整個溶膠-凝膠過程中控制顆粒的生成位點。Tamaki 等[62]利用聚苯乙烯和苯基取代硅氧烷合成了均勻的凝膠高分子材料,認為高聚物和苯基之間的 π-π 弱相互作用使得聚苯乙烯和硅酸凝膠均勻分散,而聚苯乙烯和其它不含苯基前驅體在同樣條件下制備的雜化材料表現出明顯的不均一性。這一結果說明利用芳香類高聚物結合硅氧烷的溶膠-凝膠過程制備材料時,要充分考慮芳環之間的π-π 弱相互作用。
3 展望
到目前為止,對于有機組分在分子水平上對硅氧烷水解-縮合反應過程的機理研究不是很多,因此有關該領域存在較大的研究空間。首先,結合當今各種先進的實驗分析手段和檢測技術,如液相/固相29Si NMR、SEM和TEM、SAXRD、AFM、凝膠色譜,特別是采用多種原位技術跟蹤溶膠-凝膠過程十分必要。通過29Si NMR 在線檢測可以獲得水解-縮聚反應的動力學過程;通過Raman 、IR光譜在線檢測也可以獲得水解-縮聚反應過程中物種變化的信息。另外,由于此研究體系具有不均性特點,因此,有關膠體與界面研究的方法如激光光散射、AFM、BET 、表面張力測定、光物理技術等是研究溶膠-凝膠過程的強有力手段。例如利用熒光探針技術進行在線跟蹤溶膠-凝膠過程能夠提供由于物種分布變化所引起微環境變化的重要信息。除上述有關實驗手段外,作為實驗的有力補充和證明手段,利用分子模擬方法也有助于人們從理論上認識溶膠-凝膠動力學過程[63-64]。再有,以特殊結構化合物作為“模型” 研究對象有助于對復雜體系機理的理解。通過分子設計合成具有“預想” 結構的目標產物,可以推測和驗證無機組分與有機組分相互作用的本質。我們相信,研究有機修飾對硅氧烷溶膠-凝膠過程的影響無論對于理論研究和實際應用均具有重要的意義。
參考文獻
[1] L L Hench, J K West. Chem. Rev., 1990, 90: 33~72.
[2] G Schottner. Chem. Mater., 2001, 13: 3422~3435.
[3] A V Cunliffe, S Evans, D A Tod et al. Int. J. Adhes. Adhes., 2001, 21(4): 287~296.
[4] B Alonso, D Massiot, F Babonneau et al. Chem. Mater., 2005, 17: 3172~3180.
[5] R J Hook. J. Non-Cryst. Solids, 1996, 195: 1~15.
[6] P Innocenzi , G Brusatin. Chem. Mater., 2000, 12: 3726~3732.
[7] M Gnyba, M Ker?nen, M Kozanecki et al. Opto-Electro. Rev., 2002,10(2): 137~143.
[8] L Mat?jka, O Dukh, Drahomíra et al. Macromolecules, 2001, 34: 6904~6914.
[9] Y Wei, D Jin, D J Brennan et al. Chem. Mater., 1998, 10: 769~772.
[10] K Piana, U Schubert. Chem. Mater., 1994, 6: 1504~1508.
[11] Y Kaneko, N Iyi, K Kurashima et al. Chem. Mater., 2004, 16: 3417~3423.
[12] T K Tucker, J D Brennan. Chem. Mater., 2001, 13: 3331~3350.
[13] S Yano, K Iwata, K Kurita. Mater. Sci. <st1:country-region w:st="on">Eng. C, 1998, 6: 75~90.
[14] R Winter, D W Hua, X Song et al. J. Phys. Chem., 1990, 94: 2706~2113.
[15] C A Fyfe, P P Areca. J. Phys. Chem. B, 1997, 101: 9504~9509.
[16] Y Sugahara, T Inoue, K Kuroda. J. Mater. Chem., 1997, 7(1): 53~59.
[17] M G Voronkov, V I Lavrent’yev. Top. Curr. Chem., 1982, 102: 199~236.
[18] D A Loy, B M Baugher, C R Baugher et al. Chem. Mater., 2000, 12: 3624~3632.
[19] D D Hu, C C Barghorn, M Feuillade et al. J. Phys. Chem. B, 2005, 109: 15214~15220.
[20] G A Baker, S Pandey, E P Maziarz et al. J. Sol-Gel Sci. and Technol., 1999, 15: 37~48.
[21] G Qian, Z Yang, C L Yang, J. Appl. Phys., 2000, 88(5): 2503~2508.
[22] D A Loy, K J Shea, Chem. Rev., 1995, 95: 1431~1442.
[23] 劉豐袆, 符連社, 王俊 等. 化學通報, 2003, (6): 382~387.
[24] H Muramatsu, R Corriu, B Boury. J. Am. Chem. Soc., 2003, 125: 854~855.
[25] G Cerveau, R J P Corriu, E Framery et al. Chem. Mater., 2004, 16: 3794~3799.
[26] T Koga, M Morita, H Ishida et al. Langmuir, 2005, 21: 905~910.
[27] A N Parikh, M A Schivley, E Koo et al. J. Am. Chem. Soc., 1997, 119: 3135~3143.
[28] S Vidon, R M Leblanc. J. Phys. Chem. B, 1998, 102: 1279~1286.
[29] D W Britt, V Hlady. Langmuir, 1999, 15: 1770~1776.
[30] M Popall, H Durand. Electrochimica Acta, 1992, 37: 1593~1597.
[31] F Zhang, M P Srinivasan. Langmuir, 2004, 20: 2309~2314
[32] J B Jia, B Q Wang, A Q Wu et al. Anal. Chem., 2002, 74: 2217~2223.
[33] M Abdelmouleh, S Boufi, A B Salah. Langmuir, 2002, 18: 3203~3208.
[34] X C Li, T A Ling. J. Non-Cryst. Solids, 1996, 204: 235~242.
[35] H Mori, M G Lanzendo1?rfer, A H E Mu1üller. Macromolecules, 2004, 37: 5228~5238.
[36] P Innocenzi, G Brusatin. Chem. Mater., 2000, 12: 3726~3732.
[37] S R Davis, A R Brough, A Atkinson. J. Non-Cryst. Solids, 2003, 315: 197~205.
[38] V Mellon, D Rinaldi, E B Lami et al. Macromolecules, 2005, 38: 1591~1598.
[39] J J E Moreau, L Vellutini, M Wong Chi Man et al. J. Am. Chem. Soc., 2001, 123: 1509~1510.
[40] M Barboiu, S Cerneaux, A Lee et al. J. Am. Chem. Soc., 2004, 126: 3545~3550.
[41] C J Brinker, G W Scherer. Sol-Gel Science: The Physics and Chemistry of Sol-Gel Processing. CA: Academic Press, 1990.
[42] D Phillips, P Brien, S Roberts. Sol-Gel Materials: Chemistry and Applications. <st1:country-region w:st="on">UK: Taylor & Francis Books Ltd, 2003.
[43] N Viart, J L Rehspringer. J. Non-Cryst. Solids, 1996, 195: 223~231.
[44] R F S Lenza, W L Vasconcelos. J. Non-Cryst. Solids, 2003, 330: 216~225.
[45] H K Park, T H Ha, K Kim. Langmuir, 2004, 20: 4851~4858.
[46] S P Wang, S Vidon, R M Leblanc. J. Colloid Interface Sci., 1998, 207: 303~308.
[47] D W Britt, V Hlady. J. Phys. Chem. B, 1999, 103: 2749~2754.
[48] C T Kresge, M E Leonowicz, W J Roth et al. Nature, 1992, 359: 710~712.
[49] B Hatton, K Landskron, W Whitnall et al. Acc. Chem. Res., 2005, 38: 305~312.
[50] J S Beck, J C Vartuli, W J Roth et al. J. Am. Chem. Soc., 1992, 114:10834~10843.
[51] T Amatani, K Nakanishi, K Hirao et al. Chem. Mater., 2005, 17: 2114~2119.
[52] H P Hentze, E Kr?mer, B Berton et al. Macromolecules, 1999, 32: 5803~5809.
[53] J H Jung, Y Ono, K Sakurai et al. J. Am. Chem. Soc., 2000, 122: 8648~8653.
[54] P T Tanev, Y Liang, T J Pinnavaria. J. Am. Chem. Soc., 1997, 119: 8616~8624.
[55] M Mureseanu, A Galarneau, G Renard et al. Langmuir, 2005, 21: 4648~4655.
[56] K Sahu, D Roy, S K Mondal et al. J. Phys. Chem. B, 2004, 108: 11971~11975.
[57] Q M Zhang, K Ariga, A Okabe et al. J. Am. Chem. Soc., 2004, 126: 988~989.
[58] D H Chen, Z Li, C Z Yu et al. Chem. Mater., 2005, 17: 3228~3234.
[59] A Friggeri, O Gronwald, K J C van Bommel et al. Chem. Commun., 2001, 2434~2435.
[60] 孫繼紅, 鞏雁軍, 王樹國 等. 無機化學學報, 2000, 16(1): 131~135.
[61] Y A Shchipunov, T Yu Karpenko. Langmuir, 2004, 20: 3882~3887.
[62] R Tamaki, K Samura, Y Chujo et al. Chem. Commun., 1998, 1131~1132.
[63] J S?ef ?ík, S E Rankin. J. Phys. Chem. B, 2003, 107: 52~60.
[64] H C Lay, M J S Spencer, E J. Evans et al. J. Phys. Chem. B , 2003, 107: 9681~9691.
注:本文為提供者整理翻譯的,由于知識所限,錯誤在所難免,敬請原諒。如有問題可以查找原文。
快速導航
化學品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
關于物競
物競數據庫是一個全面、專業、專注,并且免費的中文化學品信息庫,為學生、學者、化學品研究機構、檢測機構、化學品工作者提供專業的化學品平臺進行交流。
數據庫采用全中文化服務,完全突破了中英文在化學物質命名、化學品俗名、學名等方面的差異,所提供的數據全部中文化,更方便國內從事化學、化工、材料、生物、環境等化學相關行業的工作人員查詢使用。
關注我們

-
微信賬號:物競化學品數據庫

-
微博賬號:wjhxp
聯系我們
上海市延長路149號上海大學科技園412室
公司總機: 021-56389801
訂購電話: 4007001514
傳真電話: 021-56389802
客服電話: 021-56332350
電子郵件: wingch@basechem.org
 滬公網安備 31010602001115號
滬公網安備 31010602001115號