用于對映體拆分、絕對構(gòu)型的測定和質(zhì)譜測定對映體過量的新型強效手性酸
- 2012-06-04
- 專題
1.前言
現(xiàn)在大家一致認(rèn)為分子的手性是生命過程中必不可少的部分,還一致同意大多數(shù)控制活生物體生理機能的生物活性化合物都是具有手性的。因而,在針對生物活性化合物以及天然產(chǎn)物的結(jié)構(gòu)研究上,其絕對構(gòu)型的測定成為首要的重大問題。其次是成為醫(yī)藥目標(biāo)的生物活性化合物和天然產(chǎn)物的手性合成,以及如何有效的利用100%對映體純度和對映體過量(% ee)來合成所需的對映體。更深層次的講是研究手性功能分子和分子機器,例如我們實驗室開發(fā)研制的光動力手性分子馬達(dá)在近幾年發(fā)展比較迅速。因此,在材料科學(xué)領(lǐng)域,手性分子絕對構(gòu)型的準(zhǔn)確測定與絕對構(gòu)型在手性合成中的應(yīng)用一樣極其重要。
我們最近開發(fā)研制的手性羧酸作為新型手性助劑,已被證明是對映體拆分強有力的分子工具并且能明確的測定各種醇的絕對構(gòu)型。這些手性酸對于采用100%對映體過量簡便合成對映體分子是非常有用的,對其絕對構(gòu)型的分配也有著重要的作用。采用這些手性羧酸的各種合成方法已被成功的應(yīng)用于各種化合物的合成,本文稿詳盡地解釋了其基本合成方法和應(yīng)用。
2.絕對構(gòu)型的確定方法及評價
確定手性化合物的絕對構(gòu)型的方法主要分為以下兩類:
(1)確定手性化合物絕對構(gòu)型的非經(jīng)驗方法
該類別中的方法包括通過X射線晶體學(xué)的Bijvoet手性方法1)和圓二色譜激子手性方法2)。這兩種功能強大的方法可以在無需知道相應(yīng)的參考化合物絕對構(gòu)型的前提下提供目標(biāo)分子構(gòu)型的非經(jīng)驗測定。對于X射線晶體學(xué),因為重原子的不規(guī)則色散效應(yīng)在適當(dāng)條件下可以得到非常準(zhǔn)確的測定,所以我們可以依此獲得清晰而明確的絕對立體結(jié)構(gòu)。因為分子可以采用三維立體結(jié)構(gòu)的形式被投射出來以至于該方法如今被廣泛的采用。不過,X射線方法需要一個合適大小的單一晶體來適用于X射線衍射,所以此方法的關(guān)鍵問題在于如何獲得這樣的單晶。因此,采用這種方法的研究通常會變成一個漫長的試驗以及對于一種理想單晶的錯誤搜尋。
圓二色譜激子手性方法2)與前一個方法同樣的重要,是因為絕對構(gòu)型可以采用非經(jīng)驗方法確定并且不需要結(jié)晶過程。再者可用圓二色譜追蹤手性化學(xué)反應(yīng)和生物反應(yīng),甚至還可以采用這種方法來獲得不穩(wěn)定化合物的絕對構(gòu)型和構(gòu)象。然而,由于一些化合物并不是適用于這種方法的理想對象,所以所獲得結(jié)果必須仔細(xì)地加以解釋。
(2)采用已知構(gòu)型化合物作內(nèi)參比測定絕對構(gòu)型的相對方法
通過以已知絕對構(gòu)型的一個參比化合物或一個取代基作對比來測定感興趣位點上的相對構(gòu)型,從而可以獲得目標(biāo)化合物的絕對構(gòu)型。一個典型的例子就是向試樣中引入已知絕對構(gòu)型的手性輔助劑后再采用X射線晶體學(xué)確定其絕對構(gòu)型3-6)。在這種情況下,根據(jù)所引入作為內(nèi)參比物輔助劑的手性就可以自動確定有問題的這一點的絕對構(gòu)型。因而,所用樣品不需要包含有帶反色散效應(yīng)的重原子。
甚至因單晶質(zhì)量低劣而引起最終的R值不是足夠小,我們也能獲得非常清晰的結(jié)果。即便僅僅獲得了相對構(gòu)型也可以確切的確定其絕對構(gòu)型。目前已經(jīng)形成了很多種將內(nèi)參比物連接到目標(biāo)分子上的方法,例如,有離子鍵諸如傳統(tǒng)的酸堿鹽類,共價鍵諸如酯類或酰胺類以及近來研究開發(fā)的包合物的使用7-9)。這類相對的X射線方法有望得到更為廣泛的應(yīng)用。
近年來,氫質(zhì)子核磁共振(1H NMR)各向異性方法已經(jīng)經(jīng)常被用作相應(yīng)的方法來確定絕對構(gòu)型,同時在研究天然產(chǎn)物的絕對構(gòu)型上有著重要的作用。尤其是經(jīng)常采用Kusumi 等人10-13)開發(fā)的改進(jìn)Mosher方法確定仲醇的絕對構(gòu)型。如此看來,如果已知手性輔助劑[Mosher試劑α-甲氧基-α-(三氟甲基)苯乙酸(MTPA)]和[Trost試劑α-甲氧基苯乙酸]的絕對構(gòu)型,那么就可以自然而然的知道由手性仲醇與α-甲氧基-α-(三氟甲基)苯乙酸或α-甲氧基苯乙酸反應(yīng)生成的酯的絕對構(gòu)型。再者,由于在外部磁場的誘導(dǎo)下產(chǎn)生環(huán)電流致使芳香取代基(苯基)產(chǎn)生磁各向異性效應(yīng),朝著優(yōu)勢構(gòu)象中苯基的醇基部分的核磁共振信號移動到一個更高的磁場(稱為高場移)。通過觀察氫質(zhì)子核磁共振各向異性效應(yīng)來確定醇組分的絕對構(gòu)型。由于其不需要化合物結(jié)晶且核磁共振機器很容易測定所以這種方法非常方便,不過此方法的一個缺點是它是基于在溶液中分子的優(yōu)勢構(gòu)象的假設(shè),而優(yōu)點是測定非常可靠因為此方法本身有著自診斷功能。該方法已廣泛地應(yīng)用于仲醇構(gòu)型的測定,更有望擴(kuò)展到其他類型化合物構(gòu)型的測定上。
絕對構(gòu)型的相對確定還可以采用化學(xué)相關(guān)法,光學(xué)旋光[α]D比較法或者已知絕對構(gòu)型參考物的圓二色譜法。雖然這種方法也被經(jīng)常采用,但是仔細(xì)挑選參考物對于一個可靠分析來說是非常必要的。

3.手性合成方法及其評價
確定了手性化合物絕對構(gòu)型后的主要任務(wù)就是要合成該手性化合物。合成手性化合物切實可行的方法大致可以分為兩類,且每一類還可再分;這些合成方法的優(yōu)缺點如下文所述。本文中手性合成主要包括:所謂的不對稱合成和對映體拆分。此外,該方法首先采用手性輔助劑形成一個共價鍵合的非對映異構(gòu)體,然后由高效液相色譜分離最后將目標(biāo)化合物修復(fù),在廣泛意義上也被定義為對映拆分。
(1)外消旋體的對映拆分
類型a). 在這個方法中,手性輔助劑先離子鍵合到外消旋體上形成非對映異構(gòu)體混合物,然后經(jīng)分步重結(jié)晶獲得對映異構(gòu)體化合物。這種方法也適用于例如氫鍵結(jié)合7-9)的包合物的形成,關(guān)鍵點是能否用100%對映體純度通過分步重結(jié)晶來獲得非對映體。值得注意的是重結(jié)晶并非經(jīng)常能獲得100%對映純的非對映體,如果此方法成功了,它便適合大規(guī)模的制備手性化合物。
類型b). 在該方法中手性輔助劑先共價鍵合到外消旋體上生成非對映體混合物,混合物再經(jīng)硅膠高效液相色譜或其他方法分離出成對映純非對映異構(gòu)體,然后手性輔助劑裂解下來(圖1)。該方法也能產(chǎn)生一種對映體化合物,問題是能否通過高效液相色譜把這些非對映異構(gòu)體有效地分離,如果可以有效的分離開那么所獲得每一種非對映體都是對映純化合物,手性輔助劑裂解后所得到的目標(biāo)化合物也是100%對映純。最好的做法是采用一個能夠輕松裂解的手性輔助劑。
類型c). 此方法非常好地其采用了帶有手性柱(以手性固定相制成的色譜柱)的高效液相色譜或氣相色譜直接對外消旋體進(jìn)行對映體分離,這類已經(jīng)有很多研究報道過了14)。其關(guān)鍵問題也是能否將外消旋體分離成兩種對映體,如果能有效的分離那么可以通過此方法獲得100%純的對映體。該方法比較簡便適合用于分析分離,特別是因為它不需要衍生過程。總的來說手性柱都比較昂貴,大多用于分析,不過在一些情況下,工業(yè)上獲得手性化合物大多采用大規(guī)模分離如醫(yī)藥分離等。通過洗脫順序來確定絕對結(jié)構(gòu)時必須仔細(xì)分析,因為會有不少異常情況出現(xiàn)。
類型d). 此方法比較特別,因為外消旋體經(jīng)過酶或不對稱反應(yīng)的動力學(xué)拆分效應(yīng)產(chǎn)生對映體。特別是酶反應(yīng)的高立體選擇性可以產(chǎn)生高對映體純度對映體15),然而應(yīng)該小心的是該方法并不是總產(chǎn)生100%對映體純度。
(2)不對稱合成
類型a). 這是一個高效的、功能性強的合成方法,該方法通過手性試劑或手性催化劑在非手性化合物上反應(yīng)來獲得手性產(chǎn)物。作為一個眾所周知的合成方法,許多著名的綜述報道了這些不對稱合成,也沒有必要再做進(jìn)一步的解釋。但是,該方法的缺點是其獲得的產(chǎn)物并不都是對映純。進(jìn)一步講,若是基于反應(yīng)機理便很難確定產(chǎn)物的絕對構(gòu)型。因此,我們建議采用以上描述的方法來獨立地確定絕對構(gòu)型。
類型b). 還有另外一種獲得手性化合物的方法例如通過非手性或內(nèi)消旋化合物上的酶反應(yīng)生成手性化合物。通過酶催化內(nèi)消旋化合物的不對稱合成反應(yīng)尤其有趣,并被定義為去對稱化反應(yīng)。在此情況下,對映體純度也不能達(dá)到100%,必須分別確定絕對構(gòu)型。
4.用于高效液相色譜對映拆分與X-射線晶體學(xué)的強功能手性輔助劑:羧酸的應(yīng)用
實驗室制備適量的具有100%對映體純度的各種手性化合物,對映拆分1b)被認(rèn)為是一種高效的方法,如圖1所示。本方法中一種手性輔助劑共價鍵合到外消旋體上生成非對映體混合物,然后再經(jīng)硅膠高效液相色譜分離。如果色譜圖上顯示了基線分離,說明所獲得的非對映體是對映純的。該方法的特點是即使在少量情況下也能清晰、有效地分離,類型1a)描述了其與分步重結(jié)晶的對比。
我們用硅膠高效液相色譜實驗了含氮非對映體化合物(如腙16)、酰胺衍生物17))的分離。硅膠高效液相色譜能非常有效地分離手性胺與外消旋羧酸反應(yīng)生成的酰胺類非對映異構(gòu)體。然而,由于酰胺鍵不容易水解,羧酸的回收是要解決的問題。因此,需要一些額外的反應(yīng)如水解前的亞硝化反應(yīng)等17)。
我們還發(fā)現(xiàn)(1S,2R,4R)-(-)樟腦磺內(nèi)酰胺(5)作為一種手性輔助劑被成功地應(yīng)用在不對稱合成上,對各種羧酸的對映拆分反應(yīng)也至關(guān)重要。所獲得的樟腦磺內(nèi)酰胺類非對映體混合物能很好的用硅膠高效液相色譜進(jìn)行分離,再者分離后的非對映體樟腦磺內(nèi)酰胺鍵能立即被LiAlH4還原裂解生成伯醇,隨后通過氧化迅速轉(zhuǎn)化成羧酸對映體。
我們進(jìn)一步發(fā)現(xiàn)由2,10-樟腦磺內(nèi)酰胺(5)制得的酰胺一般都有良好的結(jié)晶度,很可能能夠產(chǎn)生X-射線晶體學(xué)所必需的大單晶3-6)。一般來說,需要進(jìn)行一系列的實驗來獲得能夠立即產(chǎn)生單晶的衍生物,通過此對映拆分方法可以同時獲得兩種非對映體,也提供了兩種生成單晶的可能性。如果能成功的用X-射線分析一種非對映體,那么另外一種非對映體的絕對構(gòu)型也就自然而然的確定了。
由于手性輔助劑(1S,2R,4R)-(-)2,10-樟腦磺內(nèi)酰胺(5)(TCI,東京化成工業(yè)株式會社)是由天然的(1R,4R)-(+)-樟腦油18)合成而來,所以已經(jīng)知道了它的絕對構(gòu)型。因而,它可以用來作為一個通過X-射線晶體學(xué)確定絕對構(gòu)型的內(nèi)參比物,也可以確定羧基部分的絕對構(gòu)型。2,10-樟腦磺內(nèi)酰胺(5)含有一個硫原子作為重原子,使它能夠采用Bijvoet方法來確定其絕對構(gòu)型。因此,可以采用兩種方法分別確定其絕對構(gòu)型。
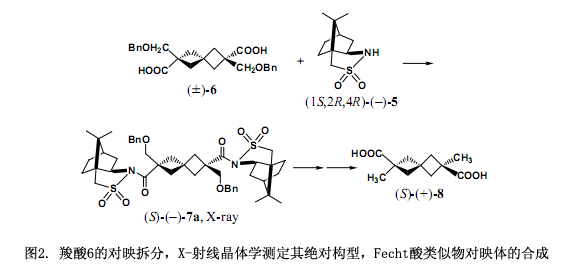
一個典型的例子如圖2所示3),(1S,2R,4R)-(-)2,10-樟腦磺內(nèi)酰胺(5)用NaH處理后生成一個陰離子,然后與酸性氯化物(±)-2,6-二(芐氧基甲基)螺[3,3]庚烷-2,6-二羧酸(6)反應(yīng),生成兩個非常容易分離的非對映體7a和7b,并在5cm的薄層色譜硅膠板上出現(xiàn)兩個明顯的黑斑(?Rf = 0.12)。在我們的實驗中單用硅膠(22φ × 300 mm: 苯/乙酸乙酯= 20:1): Rs =2.9高效液相色譜分離了大約100mg。
第一部分(–)-7a通過乙酸乙酯的重結(jié)晶產(chǎn)生柱狀晶體,然后采用X-射線晶體學(xué)分析單晶,使用樟腦部分作為一個內(nèi)參比物明確地測定其絕對構(gòu)型為S。此外,由重原子反常色散效應(yīng)所確定的絕對構(gòu)型與內(nèi)參比方法所測定的完全一致。采用LiAlH4還原(S)-(–)-7a斷裂掉手性輔助劑后,經(jīng)過幾步反應(yīng)轉(zhuǎn)變成(S)-(+)-2,6 -二甲基螺[3.3]庚烷-2,6-二元酸(8)。這樣,我們合成了非消旋體和Fecht酸類似物8的對映體,并確定了其絕對構(gòu)型3)。
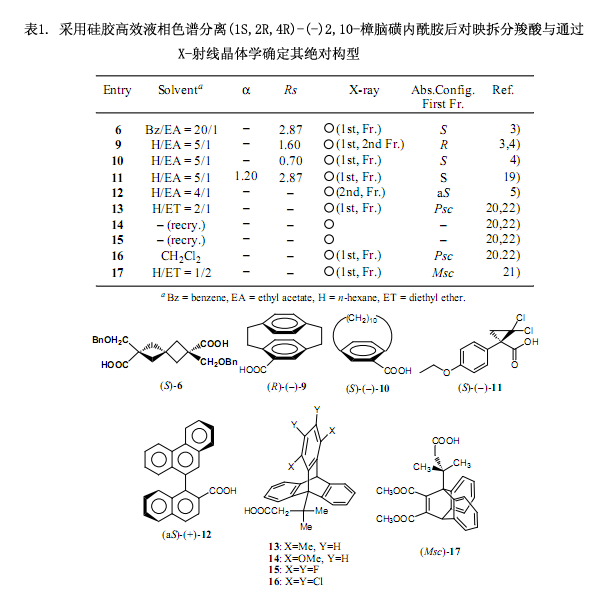
其它的應(yīng)用例子如圖1所示,該方法應(yīng)用于具有不同結(jié)構(gòu)的多種羧酸的對映拆分,因為其顯示出較好的結(jié)晶度,所以用X-射線分析能很容易確定其絕對構(gòu)型。個別化合物絕對構(gòu)型的研究在表1中列出,并揭示了一些有趣的事實。例如,曾經(jīng)通過與已知絕對構(gòu)型化合物的化學(xué)相關(guān)性確定[8] 二聚二甲苯類-10-羧酸的絕對構(gòu)型為(S)-(+)型。然而,用我們的方法4)證明此種構(gòu)型是錯誤的,我們得出的結(jié)論是化合物9和10的構(gòu)型也應(yīng)該加以修訂。盡管采用化學(xué)相關(guān)性所確定的絕對構(gòu)型被認(rèn)為是最可靠的方法,但是像這樣的錯誤很容易發(fā)生,所以在使用時應(yīng)多加注意。
由Toyota等人研究的化合物13-17也是具有旋轉(zhuǎn)對映異構(gòu)性和光學(xué)活性物質(zhì)的有趣例子。總的來說,基于空間位阻旋轉(zhuǎn)對映異構(gòu)現(xiàn)象很難確定手性化合物的絕對構(gòu)型,不過Toyota等人采用上述方法成功的解決了這個問題20-22)。
5.用于高效液相色譜法和X-射線晶體法分析醇類對映拆分的高效手性輔助劑的分子設(shè)計
上文已經(jīng)詳細(xì)敘述的手性助劑2,10-樟腦磺內(nèi)酰胺(5)在對映拆分以及羧酸絕對構(gòu)型的確定上至關(guān)重要,然而,對于手性醇合成以及確定其絕對構(gòu)型的需求遠(yuǎn)大于羧酸。關(guān)鍵的問題是在保留上述手性助劑特點的同時要采用什么樣的助劑來適用于這類醇。
我們采用以下的分子設(shè)計來處理這個問題,首先,設(shè)計引入了連接2,10-樟腦磺內(nèi)酰胺和醇的連接物(圖3)23),2,10-樟腦磺內(nèi)酰胺的一個酰胺鍵和醇的一個酯鍵選擇性鍵合到連接物上,由于酰胺鍵跟硅膠高效液相色譜親和力較強而被保留下來,而酯鍵很容易轉(zhuǎn)變以及裂解掉常被用來生成醇。因此,鄰苯二甲酸作為連接分子的首選23),在對苯二甲酸和琥珀酸上兩個手性部分在空間上被分割開來。但是,在鄰苯二甲酸上兩個手性部分比較接近以至于產(chǎn)生更強的相互作用。我們推斷高效液相色譜能有效地識別其非對映體。
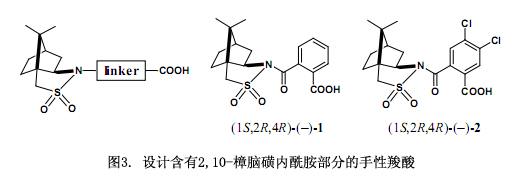
目標(biāo)物手性鄰苯二甲酸(–)-1是由(1S,2R,4R)-(-)2,10-樟腦磺內(nèi)酰胺(5)陰離子與鄰苯二甲酸酐反應(yīng)所合成的,在CHCl3中的熔點為184-187℃;[α]D20 -134.7 (c 2.218, 甲醇)。化合物1應(yīng)正式稱為手性鄰苯二甲酸酰胺,不過在這里我們采用其通用名手性鄰苯二甲酸。在二環(huán)己基碳二亞胺(DCC)和4-二甲氨基吡啶(DMAP)存在條件下,用醇來縮合該羧酸23)。
以下例證詳述了制備步驟,手性鄰苯二甲酸(-)-1可以與(±)-α-甲基-(4 - 溴芐基)甲醇(18)(表2所示)反應(yīng),隨后用硅膠高效液相色譜(正己烷/乙酸乙酯= 3:1; α = 1.1, Rs = 1.3.)分離所生成的非對映體混合物。如果分離因數(shù)α大于等于1.1,那么所生成的化學(xué)組分通常是可以完全分離。酯19b作為第二組分被洗脫出來而且具有良好的結(jié)晶度,經(jīng)過在甲醇溶液中的重結(jié)晶后生成適合于X-射線分析的單晶。從X-射線掃描的ORTEP圖上看出,通過采用2,10-樟腦磺內(nèi)酰胺部分作為內(nèi)部參考物以及采用重原子效應(yīng)測定醇部分的絕對構(gòu)型,可以清晰地將其確定為S型。還從非對映體酯19b中重新獲得了對映體醇(S)-(–)-1823)。
如表2所示的其他例子,我們成功的對映拆分了各種醇并采用X-射線分析確定了其絕對構(gòu)型,表中所列舉回收的對映純醇再次證明了此方法在合成上的有效性。在氰醇27和胺28中可以采用這種方法進(jìn)行非對映體分離和絕對構(gòu)型的確定。不過,存在的問題是對映純化合物27與28如何回收,原因是酰胺鍵不容易水解掉。酰胺28作為(2R,3R)-(+)-酒石酸19)的鹽被對映分解,通過采用此方法確定了其絕對構(gòu)型是(S)-(–)型。對于化合物29即使在它的結(jié)晶度較差所導(dǎo)致R值比較小的情況下,也可以通過采用內(nèi)參比方法清晰的確定其絕對構(gòu)型。
在我們研究的過程中,我們發(fā)現(xiàn)手性鄰苯二甲酸(–)-1的酯類通常具有較差的溶解性,可能是因為其極易結(jié)晶而導(dǎo)致其在硅膠高效液相色譜里洗脫時間延長。再者就是在重結(jié)晶過程中生成細(xì)微針形的晶粒致使其不適合用X-射線晶體學(xué)分析。這說明使用低溶解性的手性鄰苯二甲酸酯具有一定的反作用,因此我們在探索一系列的化合物中應(yīng)該尋找能生成具有較強溶解性的軟酯連接物。
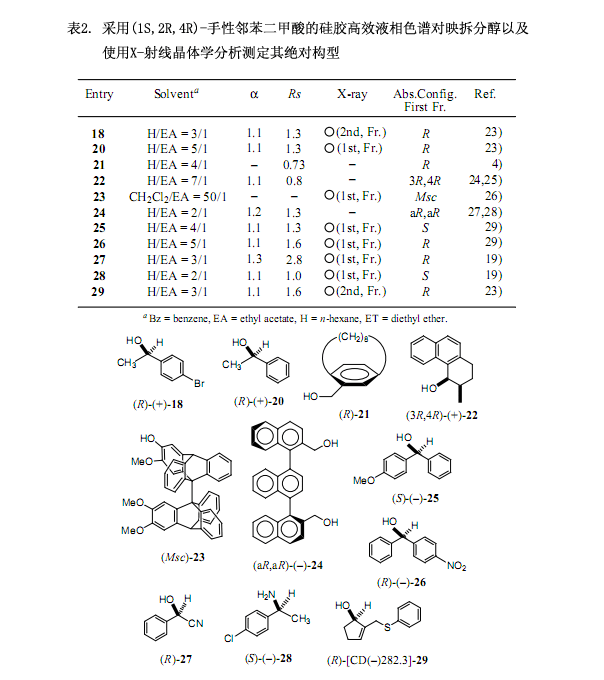
6.較強功能手性二氯代鄰苯二甲酸用于醇的高效液相色譜對映拆分和X-射線晶體分析:發(fā)展與應(yīng)用
經(jīng)過一系列的調(diào)查研究,我們發(fā)現(xiàn)手性二氯代鄰苯二甲酸(–)-2[在乙醇中的熔點221℃;[α]D20 –101.1 ;(c 1.375, 甲醇);圖3]是由4,5-二氯代鄰苯二甲酸作連接物而制備,有效地解決了上述討論的問題,也就是其具有較高的溶解性能和在高效液相色譜中的洗脫時間比較短。并且,還能提供適用于X-射線分析的柱狀晶體。例如對于表2及圖4中的(±)-順式-1,2,3,4 -四氫-3-甲基-4-菲酚(22)來說,采用手性鄰苯二甲酸洗脫這兩個非對映體時間分別為27.6分鐘和31.0分鐘,但是在同樣的條件下采用手性二氯代鄰苯二甲酸洗脫的時間分別是14.6和16.7分鐘,后者所用的時間是前者的一半左右,更進(jìn)一步說就是提高了其分離和分配因子24)。
手性輔助劑二氯代鄰苯二甲酸(–)-2是一種重要的內(nèi)參比物在采用X-射線分析測定絕對構(gòu)型時,跟手性鄰苯二甲酸(–)-1作用相似。其中,羧酸2除了含有一個硫原子外還含有2個氯原子作為重原子,因而通過重原子的反常色散效應(yīng)可以有效地測定絕對構(gòu)型。
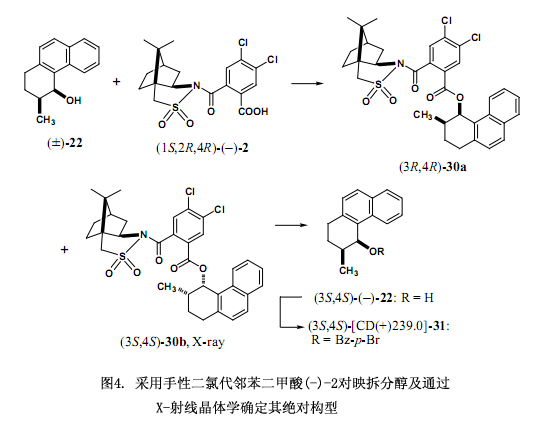
如圖4所示的例子醇(±)-22在二環(huán)己基碳二亞胺(DCC)和4-二甲氨基吡啶(DMAP)條件下用手性二氯代鄰苯二甲酸(–)-2濃縮后獲得兩種酯的非對映體混合物,混合物再經(jīng)過硅膠高效液相色譜[正己烷/乙酸乙酯=7:1,α = 1.18, Rs = 1.06]分離。酯30a在甲醇中重結(jié)晶后洗脫出的第一組分生成柔滑細(xì)微針形晶粒,不適于用X-射線分析,第二組分30b在乙醇中重結(jié)晶后生成較大的棱狀晶體可用X-射線分析。采用2,10-樟腦磺內(nèi)酰胺作內(nèi)參比物以及用重原子效應(yīng)都能清楚的確定30b的絕對構(gòu)型是(3S,4S)型。LiAlH4還原酯30b來去掉手性輔助劑后生成對映純醇(3S,4S)-(–)-2224,25),這種方法所測定的絕對構(gòu)型與采用圓二色譜激子手性方法測定的結(jié)果相一致,這些方法也適用于測定相應(yīng)的4 - 溴苯甲酸(31)的絕對構(gòu)型24)。
近來我們還發(fā)現(xiàn)使用K2CO3/甲醇溶劑分解可以有效地從手性二氯代鄰苯二甲酸酯獲得對映純醇,并具有較高的產(chǎn)率。
表3列舉了其他一些應(yīng)用實例,即使對位取代物決定著遠(yuǎn)離手性中心(如羧基上的碳原子)分子的手性,但是手性二氯代鄰苯二甲酸酯類、對位取代二苯甲醇類25,26與34-38仍可以被硅膠高效液相色譜有效的分離29,30,32)。這些結(jié)果表明手性二氯代鄰苯二甲酸很容易識別分子的手性,例如氫原子與對位取代官能團(tuán)的差別。
手性識別(4-甲基苯基)-苯甲醇(36)中的醇官能團(tuán)對于手性二氯代鄰苯二甲酸酯就是不大可能的,因為手性分子中氫原子與甲基的差別不大,所以不能識別其手性。因而也無法利用硅膠高效液相色譜來分離非對映體。需要采用以下步驟來解決該問題,第一步:選擇(4-溴苯基)(4'-甲苯基)甲醇(37)作為前驅(qū)體,用手性二氯代鄰苯二甲酸對其進(jìn)行對映分解,然后利用X-射線晶體學(xué)測定他們的絕對構(gòu)型。把溴原子還原生成所要的對映純醇(S)-(–)-3629),這一步是合成的關(guān)鍵,同位素取代的手性二苯甲醇的絕對構(gòu)型的測定在下文敘述。
同位素取代物也可以產(chǎn)生分子手性,如2H取代二苯甲醇39上的氫原子,13C取代二苯甲醇43上的碳原子32,33)。不能直接用普通手性固定相高效液相色譜或者帶有手性助劑硅膠高效液相色譜來對映分解這些同位素取代的手性化合物。諸如這個例子,首先必須選擇一種前驅(qū)體如(±)-(4-溴苯基)(苯基-2,3,4,5,6-d5)甲醇(40),然后對映拆分二氯鄰苯二甲酸并同時測定其絕對構(gòu)型。既而將溴原子還原生成所需要的對映純(苯基-2,3,4,5,6-d5)苯甲醇(39)32)。

利用以下步驟制備(苯基-2,3,4,5,6-d5)苯甲醇(39),采用手性二氯代鄰苯二甲酸跟外消旋醇(±)-40縮合反應(yīng),隨后采用硅膠高效液相色譜分離所生成的酯。經(jīng)過一系列的重結(jié)晶過程兩種非對映體酯都能生成針形晶體,先從第一組分41a回收對映純醇(–)-40,另外還有部分(–)-40經(jīng)樟腦酸性氯化物處理后轉(zhuǎn)變成酯42。酯42具有良好的結(jié)晶度,生成適于X-射線分析的柱狀晶體,采用(–)-樟腦酸的絕對構(gòu)型作為內(nèi)參比物測定醇部分的絕對構(gòu)型為S型。然后在Pd-C存在的條件下利用H2NNH2/H2O還原醇(S)-(–)-40生成同位素取代的對映純(苯基-2,3,4,5,6-d5)苯甲醇[CD(–)270.4]-(S)-39,并且可以明確的測定其絕對構(gòu)型。利用波長為589nm的鈉D線測算比旋光度[α]D來區(qū)分對映體,但是,很難測定同位素取代手性化合物的比旋光度[α]D。因此,我們提供一種采用圓二色譜數(shù)據(jù)測定對映體的新定義方法,由于圓二色譜不僅僅比比旋光度[α]D靈敏,而且在測定微量樣品的時候更為精確。例如,[圓二色(–)270.4]-(S)-39在270.4nm處表明為負(fù)的圓二色對映體,測定其絕對構(gòu)型為S型32)。
如果在一開始就使用(–)-樟腦酸作為對映拆分的手性助劑,那么這將變得更為有利,但是,(–)-樟腦酸的對映拆分能力比較弱。例如,由醇40生成的樟腦酸酯并不可以用硅膠高效液相色譜分離,因而有時依據(jù)不同的形勢有必要選擇兩種手性助劑。
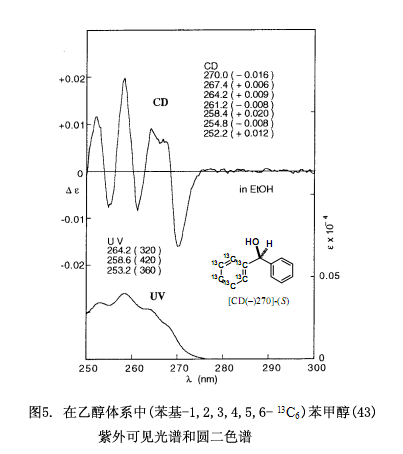
采用相似的合成步驟來合成13C同位素取代的手性二苯甲醇(43)33),也就是說,先選擇(4-溴苯基)(苯基-1,2,3,4,5,6 - 13C6)甲醇(±)-(44),隨后將其對映拆分并采用與上述描述相似的方式來測定它的絕對構(gòu)型。再將溴原子還原生成13C取代的手性(苯基-1,2,3,4,5,6-13C6)苯甲醇[圓二色(–)270]-(S)-43。盡管由于同位素12C和13C之間有細(xì)微的差別而致的非常弱的分子手性,但是卻能很容易的觀察到13C取代的手性二苯甲醇(S)-43的圓二色譜,圓二色譜圖如圖5所示33)。
對于鄰取代的二苯甲醇我們也獲得了非常有趣的結(jié)果,使用以上描述的方法制備了對映體(2 -甲氧基苯基)苯甲醇(-)-(45),并測定其絕對構(gòu)型為S型34)。以前采用不對稱催化反應(yīng)合成了手性(2-甲苯基)苯甲醇(46),基于手性反應(yīng)機理估計了其絕對構(gòu)型。但是,基于手性機理來確定的絕對構(gòu)型結(jié)果是否可靠?在這樣的疑問下就很有必要獨立的、明確的來測定絕對構(gòu)型。為了解決這個問題我們進(jìn)行了以下實驗,但不能成功的直接將46對映拆分成手性二氯代鄰苯二甲酸,即使是有鄰取代醇36存在也不行,因為很難區(qū)分鄰位上的氫原子和甲基。然后我們嘗試對映拆分(4-溴苯基(2’-甲苯基)甲醇(47),可是采用高效液相色譜分析時僅僅出現(xiàn)了一個峰。
然后我們采用以下間接方法34-36),此方法包括把(2-羥基-甲苯基)苯甲醇(48)對映拆分成手性二氯代鄰苯二甲酸,用X-射線分析測定其絕對構(gòu)型,將48的對映純衍生物轉(zhuǎn)化成所要的醇46。手性二氯代苯甲酸(–)-2(方程式1)可以與二醇(±)-48反應(yīng)生成酯非對映體混合物,這些酯主要是通過對伯醇官能團(tuán)進(jìn)行的選擇性酯化反應(yīng)而合成的。在此情況下,手性助劑官能團(tuán)與伯醇部分成鍵結(jié)合后距48的手性中心較遠(yuǎn),因而很容易分離非對應(yīng)酯。從第二部分(–)-49b可以獲得單晶,用X-射線分析測定其絕對構(gòu)型為S型。第一洗脫部分(R)-(–)-49a經(jīng)過幾步反應(yīng)轉(zhuǎn)變成所要的對映純醇(R)-(–)-46,該結(jié)果顯示先前采用基于不對稱反應(yīng)機理所確定46的絕對構(gòu)型是錯誤的35),因為基于反應(yīng)機理所確定的絕對構(gòu)型有時可能產(chǎn)生錯誤,最明智的做法是分別測定產(chǎn)物的絕對構(gòu)型。
這種方法還能有效的制備各種芐醇52-5537),這類醇還作為天然產(chǎn)物全部合成的手性合成子,原因是這些醇具有明確的絕對構(gòu)型以及100%對映體純度。
阿托異構(gòu)體24是一個特殊的手性化合物,因為它含有三個萘發(fā)色團(tuán),其對映拆分和絕對構(gòu)型的測定按下述方法執(zhí)行27,28)。外消旋二醇、(±)-1,1':4',1'' 三萘-2,2''-二甲醇(24)加成到手性二氯代苯甲酸上形成一種酯類混合物,然后用硅膠高效液相色譜(正己烷/乙酸乙酯= 2:1, α = 1.18)分離。然而第一組分(–)-56a經(jīng)正己烷/乙酸乙酯重結(jié)晶生成細(xì)微針形晶體,第二組分(+)-56b生成較大晶體。
總的來說,適用于X-射線分析的單晶必須是具有一定表面和邊緣的棱狀或者柱狀晶體,也可以是厚板形式的晶體。酯(+)-56b經(jīng)重結(jié)晶后生成具有跟飛機翅膀相似的三角形翅膀的晶體,這些晶體看起來不像單晶,將翅膀移開后剩余部分就是單晶,可以用X-射線測定主體部分的構(gòu)型。更加有趣的是,由初步晶格常數(shù)估算的不對稱單元的分子量跟(+)-56b分子量不一致,因此我們初步認(rèn)為此分子的結(jié)構(gòu)可能不正確,由手性二氯代鄰苯二甲酸的不對稱性部分可知,我們可以假設(shè)56b的不對稱部分含有一個分子。認(rèn)真考察所獲得的數(shù)據(jù)可以看出56b分子的一半等價于一個不對稱單元,也即是在酯(+)-56b晶體中有一個C2對稱結(jié)構(gòu),盡管事實上此分子中含有復(fù)雜的手性二氯代鄰苯二甲酸部分。例如利用內(nèi)參比法測定扭轉(zhuǎn)三萘生色團(tuán)的絕對構(gòu)型為(aS,aS),從酯(aS,aS)-(+)-56b上移去手性助劑生成對映純二醇(aS,aS)-(+)-24,利用X-射線分析測定的絕對構(gòu)型與采用圓二色譜激子手性方法測定的(+)-24絕對構(gòu)型相符27,28)。

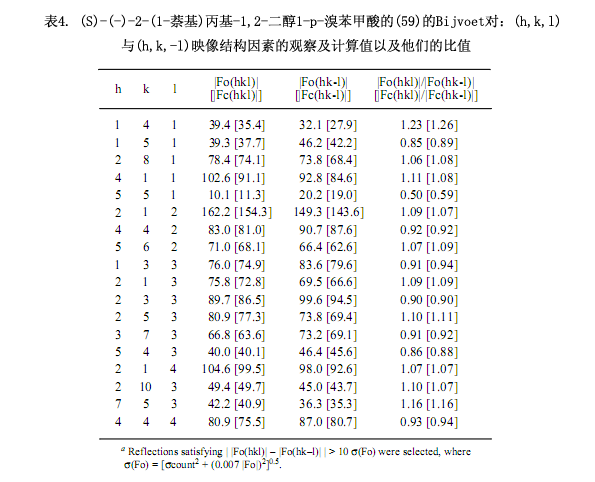
2-(1-萘基)丙基-1,2-二醇化合物作為在家兔體內(nèi)1-異丙基萘的手性代謝產(chǎn)物而被分離,該代謝產(chǎn)物不是對映體,依據(jù)反應(yīng)機理僅能憑經(jīng)驗估計其絕對構(gòu)型。為了獲得對映純二醇57以及用一種清晰的方法測定其絕對構(gòu)型,手性二氯代鄰苯二甲酸方法可以用于(±)-57絕對構(gòu)型的測定38)。在這種情況下,只有伯醇部分被酯化后得到非對映體混合物,再用硅膠高效液相色譜(正己烷/乙酸乙酯 = 4:1, α = 1.3, Rs = 1.1.)分離此混合物。在此高效液相色譜中自由叔醇官能團(tuán)的存在尤為重要,但是對叔醇官能團(tuán)的保護(hù)致使分離更加困難。
即使不斷地重結(jié)晶,但是所得到的兩種非對映體僅是無定形固體。用LiAlH4還原第一部分(–)-58a生成對映純乙二醇(–)-57,然后進(jìn)一步轉(zhuǎn)化成4 - 溴苯甲酸(–)-59(圖6.)。由乙醇對其重結(jié)晶,(–)-59能生成適合于X-射線分析的單晶,通過對含有溴原子的反常色散效應(yīng)的Bijvoet對測量可以確切的確定其絕對構(gòu)型為S型(表4)。
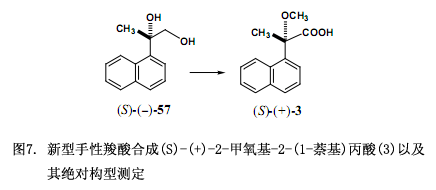
進(jìn)一步說,我們通過幾個反應(yīng)步驟從二醇(S)-(–)-57獲得對映體(S)-(+)-2-甲氧基-2-(1-萘基)丙酸(3)(圖7)38)。如下討論我們發(fā)現(xiàn)對對映拆分和絕對構(gòu)型測定非常有效的新型羧酸39-41)。我們針對手性鄰苯二甲酸(1)與二氯代鄰苯二甲酸(2)應(yīng)用研究與該文獻(xiàn)(J. Synth.Org. Chem. Jpn. (Yuki Gosei Kagaku Kyokaishi)報道的相一致42)。
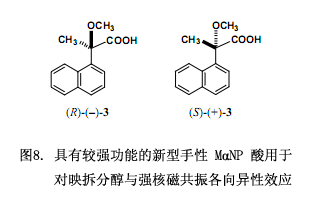
7.一種新型手性羧酸2-甲氧基-2-(1-萘基)丙酸(MαNP 酸),對醇的對映拆分與采用氫質(zhì)子核磁共振各向異性方法測定其絕對構(gòu)型的重要作用:基本原理及應(yīng)用
我們已經(jīng)探討手性鄰苯二甲酸與二氯代鄰苯二甲酸的設(shè)計和應(yīng)用,二者對于對映體化合物的合成以及采用X-射線分析明確的確定其絕對構(gòu)型有著重要作用。然而表2和表3所列舉的大部分應(yīng)用僅限于芳香族化合物絕對構(gòu)型的測定,是否有一種功能較強的方法來用于測定脂肪族化合物的絕對構(gòu)型?
我們近來還發(fā)現(xiàn)新型手性羧酸2-甲氧基-2-(1-萘基)丙酸(MαNP酸 (3)),能有效的對映拆分脂肪醇,尤其是無環(huán)脂肪醇。由MαNP 酸3跟醇類反應(yīng)生成的酯的氫質(zhì)子核磁共振譜圖可知,醇組分中的質(zhì)子化學(xué)位移受萘官能團(tuán)誘導(dǎo)的磁各向異性效應(yīng)影響強烈38-41)。因而,此羧酸3在改進(jìn)的Mosher方法中可以被用作手性助劑10),羧酸3對測定仲醇的絕對構(gòu)型也非常有用。該羧酸3的另外一個優(yōu)點就是它不具外消旋作用,因為3的α位是飽和的,以至于對映純3很容易制備。以下討論,MαNP 酸3是功能強的手性衍生劑,可以確保仲醇的對映拆分及其絕對構(gòu)型的測定同時進(jìn)行。這里說的MαNP 酸3在測定天然產(chǎn)物和合成具有生物活性的手性化合物(例如手性藥物)的絕對構(gòu)型時作用較強。從這種意義上來看,手性MαNP 酸3優(yōu)于傳統(tǒng)的手性酸Mosher’s MTPA酸10),Trost’s MPA酸12)以及由Riguera11)and Kusumi10)團(tuán)隊研發(fā)的1- and 2-NMA酸。
以下部分詳細(xì)描述手性MαNP 酸3的基礎(chǔ)理論和應(yīng)用:手性酸3的合成;由X-射線與化學(xué)相關(guān)法測定其絕對構(gòu)型;通過手性醇對映拆分外消旋酸3以及使用手性醇確定其絕對構(gòu)型;采用核磁共振法與圓二色譜光譜法測定MαNP 酸酯的絕對構(gòu)型和構(gòu)象分析;用手性MαNP 酸3對醇對映拆分以及確定醇的絕對構(gòu)型;從分離的非對映酯回收100%對映體純度的手性醇;該方法應(yīng)用于各種醇。
8.MαNP 酸3的簡便合成,與天然(-)-薄荷醇結(jié)合后顯著的對映拆分作用
為了合成大量對映純手性8. MαNP 酸(3),圖9顯示了外消旋酸3的簡便合成和對映拆分過程。總而言之,手性合成的胺或生物堿就是用于對映拆分羧酸,我們采用下面的新方法應(yīng)用于手性醇,在此方法中用外消旋酸3來縮合手性醇,形成的非對應(yīng)酯采用硅膠高效液相色譜分離,分離后的酯再經(jīng)水解生成對映體和目標(biāo)羧酸。
自然狀態(tài)的薄荷醇作為一種手性醇而被選擇,用外消旋酸3酯化該醇,我們發(fā)現(xiàn)很容易生成非對映酯63a與63b,再經(jīng)硅膠高效液相色譜(正己烷/乙酸乙酯 =10:1)分離,其過程如圖10所示。分離因數(shù)與分離度都很大(α= 1.83, Rs = 4.55),這同時還表明酸3有很強的識別醇手性的能力。酯63a先洗脫出來受到溶劑分解作用生成手性酸(+)-3,而63b隨后被洗脫出生成酸(–)-3。為了測定所獲得手性酸3的絕對構(gòu)型先將其轉(zhuǎn)化成甲酯,再用圓二色譜測定。把這些圓二色譜圖跟X-射線分析與化學(xué)相關(guān)法測定的真實樣品的絕對構(gòu)型相對比,所得兩種酯的絕對構(gòu)型分別是(S)-(+) 和 (R)-(–),也就是(S)-(–)-63a和(R)-(–)-63b(圖9所示)。

9.氫質(zhì)子核磁共振各向異性方法測定仲醇的絕對構(gòu)型:扇形規(guī)則及應(yīng)用
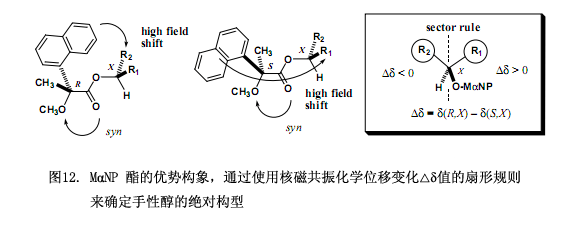
如上所述,氫質(zhì)子核磁共振各向異性方法已經(jīng)經(jīng)常被用作一種相關(guān)的驗證方法來測定手性有機化合物的絕對構(gòu)型10-13),尤其是測定手性仲醇的改進(jìn)Mosher方法已經(jīng)成功用于測定天然產(chǎn)物的絕對構(gòu)型。至于Mosher’s MTPA 酸與 Trost’s MPA 酸,其苯基呈現(xiàn)磁各向異性效應(yīng)主要是因芳環(huán)環(huán)電流所致,再引起醇中質(zhì)子的化學(xué)位移(δ)發(fā)生變化。因此,可以用(R)和(S)羧酸所形成的酯的化學(xué)位移變化(?δ)(其中?δ = δ(R) – δ(S) 或者 ?δ = δ(S) – δ(R))來確定手性醇的絕對構(gòu)型。我們發(fā)現(xiàn)MαNP 酸3要優(yōu)于Mosher’s MTPA 酸和 Trost’s MPA 酸,原因是萘基的磁各向異性效應(yīng)比苯基的磁各向異性效應(yīng)要大,進(jìn)而導(dǎo)致化學(xué)位移變化?δ值增大,因此當(dāng)采用MαNP 酸3作為手性核磁共振各向異性試劑,手性醇的絕對構(gòu)型可以清楚的確定。
再者就是MαNP 酸還有另外一個優(yōu)點即它本身不具有外消旋作用,因為它的α位上也是飽和的。從這些原因來看,在測定手性醇包括天然產(chǎn)物的絕對構(gòu)型時,最好的做法就是使用MαNP 酸3而不是用傳統(tǒng)的手性酸。
非對映體MαNP 酸酯63a和63b所有的核磁共振質(zhì)子峰通過各種方法被分配開來,其中還包括二維方法(1H, 1H-1H COSY, 13C, 1H-13C COSY, HMBC, 圖 11a)。酯63b中異丙基上的質(zhì)子跟63a中的質(zhì)子相比主要出現(xiàn)在高場區(qū),另一方面63a中2位上的質(zhì)子跟63b上的質(zhì)子相比也出現(xiàn)在高場區(qū)。這些化學(xué)位移的明顯變化主要是由于MαNP 酸中萘基的磁各向異性效應(yīng)所致。

從核磁共振各向異性效應(yīng)來確定絕對構(gòu)型,需要確定每一個非對映體的優(yōu)勢構(gòu)象。在酯63a和63b中MαNP 酸與薄荷醇的絕對構(gòu)型已由上述方法確立,下面的穩(wěn)定構(gòu)型主要是滿足從核磁共振圖譜中觀察到的各向異性效應(yīng)(圖11)。也即是在它們各自穩(wěn)定的構(gòu)型中,甲氧基跟酯羰基里面的兩個氧原子相互發(fā)生順疊,還有酯羰基氧原子順疊到醇甲烷質(zhì)子上,至此甲氧基、酯基以及醇甲烷質(zhì)子在同一平面上,該平面被稱作MαNP平面(圖11)。這些順疊構(gòu)象跟MPA酯里面的構(gòu)象相似,在酯63a中萘基和H-2質(zhì)子都位于MαNP平面的同一前側(cè),并且H-2質(zhì)子位于萘基之上,因而H-2質(zhì)子受到高場位移的磁各向異性效應(yīng),所以出現(xiàn)在高場區(qū);在酯63b中萘基與異丙基相距較近,可以觀察到異丙基質(zhì)子具有高場位移。
酯63a和63b順疊構(gòu)象的優(yōu)勢還可以通過對比其核磁共振數(shù)據(jù)跟2-羥基-2-(1-萘基)丙酸薄荷酯的核磁共振數(shù)據(jù)(圖11(b)所示)來看出。從核磁共振化學(xué)位移及紅外數(shù)據(jù)可以很明顯的看出叔醇羥基官能團(tuán)中的氫原子鍵合到酯羰基的氧原子上,也即是羥基與酯羰基氧原子產(chǎn)生順疊構(gòu)象,我們還發(fā)現(xiàn)一個有趣的現(xiàn)象2-羥基-2-(1-萘基)丙酸薄荷酯(S;1R,3R,4S)-(–)-63a的核磁共振化學(xué)位移數(shù)據(jù)尤其是薄荷醇部分的數(shù)據(jù)跟(圖11(a)、(b)所示)2-羥基-2-(1-萘基)丙酸薄荷酯(S;1R,3R,4S)-(–)數(shù)據(jù)極其相似。對其他非對映體對來說亦是如此,例如(R;1R,3R,4S)-(–)-63b2-羥基-2-(1-萘基)丙酸薄荷酯(R;1R,3R,4S)-(–)也是這樣的結(jié)果。這些事實進(jìn)一步說明MαNP酸薄荷酯呈現(xiàn)順疊構(gòu)象,HαNP酸薄荷酯也是這樣,而且這都完全可以用磁各向異性效應(yīng)來解釋。
根據(jù)采用MαNP酯核磁共振各向異性效應(yīng),我們可以引出適用于確定仲醇絕對構(gòu)型的扇形規(guī)則(圖12)。基本步驟如下:(R)-MαNP酸 和(S)-MαNP酸分別與一種手性醇反應(yīng),將其絕對構(gòu)型定義為X型,因此由(R)-MαNP酸所制得酯具有(R,X)絕對構(gòu)型,另外一個由(S)-MαNP酸制得酯絕對構(gòu)型為(S,X)型。(R,X)- 酯和(S,X)-酯所有的核磁共振質(zhì)子信號經(jīng)仔細(xì)分析后被完全分配,如有必要我們建議使用二維光譜。化學(xué)位移變化?δ值((?δ= δ(R,X) – δ(S,X))適用于計算醇中的所有質(zhì)子。圖12顯示了用于MαNP酯的扇形規(guī)則,在這里面MαNP基團(tuán)放置在前下方,而仲醇中甲烷質(zhì)子放置在后下方;帶有化學(xué)位移變化?δ正值的R1基團(tuán)質(zhì)子放置在右邊,而帶有化學(xué)位移變化?δ負(fù)值的R2基團(tuán)質(zhì)子放置在左邊。從這個投影上看,也就可以確定手性醇的絕對構(gòu)型是X型。
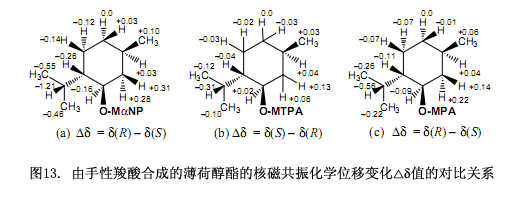
手性MαNP酸的磁各向異性效應(yīng)比常用手性羧酸的磁各向異性效應(yīng)強(圖13),例如MαNP酸薄荷酯的化學(xué)位移變化?δ值大約是Mosher’s MTPA酯的?δ值的四倍還要大10)(圖13b),大約是Trost’s MPA酯的兩倍12)(圖13c),是Riguera11)和 Kusumi10)等人報道的2-NMA酸酯的?δ值的兩倍。因此,MαNP酸酯可以有效的測定天然產(chǎn)物的絕對構(gòu)型。
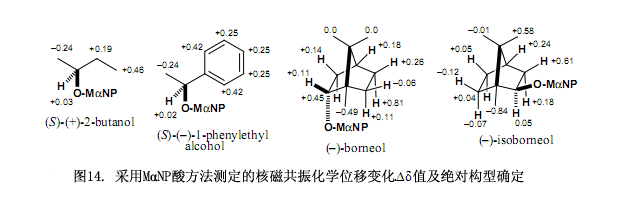
此MαNP酸方法在其他手性醇上的應(yīng)用如圖14所示。
10.使用MαNP酸對不同醇的手性拆分及其絕對構(gòu)型的同時測定41)
MαNP酸的另外一個優(yōu)點就是其具有較強的手性識別能力,例如正如以上所討論的外消旋MαNP酸可以成功的對映拆分天然(–)-薄荷醇酯。所生成的非對映酯能夠采用硅膠高效液相色譜清楚的分離,MαNP酸可以對映拆分如圖14所列出的其他手性醇,這些事實有邏輯地表明如果使用對映純MαNP酸可以對映拆分外消旋醇,事實上我們成功的運用(S)-(+)-3MαNP酸對映拆分了不同醇,例如圖15所示。

新型手性(S)-(+)-3 MαNP酸具有功能顯著對映拆分醇的能力,尤其是對映拆分手性醇。例如2-丁醇所合成的非對映酯在分離因數(shù)α = 1.15,分離度Rs = 1.18時基本上都能被分離,在此情況下手性羧酸3就可以明顯的識別僅有細(xì)微差別的甲基和乙基官能團(tuán)。因為手性酸3對脂肪醇呈現(xiàn)出很強的分辨能力所以該方法是一種優(yōu)越而又實用的方法,還可以用于幾乎不可能實現(xiàn)的的不對稱合成反應(yīng)上。
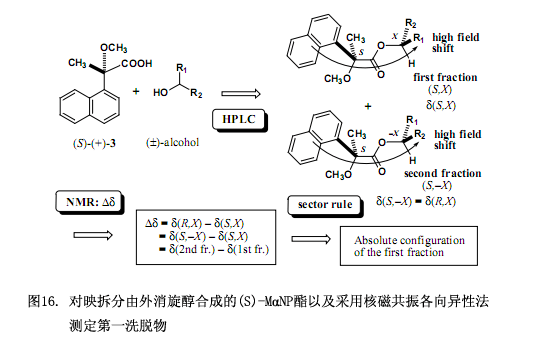
而后另外的問題是怎樣測定醇部分的絕對構(gòu)型,用上述描述的手性MαNP酸依據(jù)核磁共振各向異性效應(yīng)測定所分離的非對映體的絕對構(gòu)型。其總體方案如圖16所示,用(S)-(+)-3MαNP 酸酯化外消旋醇生成非對映酯混合物,再用硅膠高效液相色譜分離此混合物。第一洗脫物的絕對構(gòu)型被定為(S,X),這里的S表示MαNP酸部分的絕對構(gòu)型,而X表示醇部分的絕對構(gòu)型。酯第二洗脫物的絕對構(gòu)型表示為(S,–X),此處的-X為X相反的絕對構(gòu)型。化學(xué)位移變化?δ值的最初定義是?δ = δ(R,X) – δ(S,X),若要計算?δ值必須先知道δ(R,X)的值,然而此方案中不存在對映體(R,X),因此化學(xué)位移變化?δ的原始方程在這里沒有多大用處。
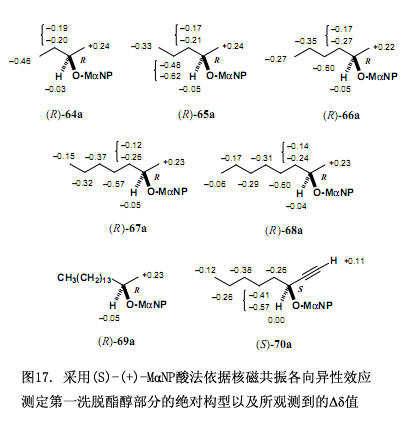
為了解決以上問題需要對方稱進(jìn)行如下改變,因為酯(S,–X)是酯(R,X)的對映體,因而他們的核磁共振數(shù)據(jù)彼此相同,也即?δ = δ(R,X) – δ(S,X)= δ(S,–X) – δ(S,X) = δ(2nd fr.) – δ(1st fr.),第一洗脫部分的絕對構(gòu)型X可以由化學(xué)位移變化?δ值來確定,此?δ值也就是用第一洗脫部分的化學(xué)位移值來減去第二洗脫部分的化學(xué)位移值而得到(圖16)。本方法已經(jīng)用于圖15所示的酯,可以得到?δ值及第一洗脫酯的絕對構(gòu)型(圖17),這些?δ值已被合理分布,右邊是正值,左邊是負(fù)值,因而可以確定第一洗脫酯的絕對構(gòu)型,相反的絕對構(gòu)型就是第二洗脫酯的了。所需要注意的是如果采用(R)-(–)-3MαNP 酸,那么就應(yīng)該這樣定義?δ值,?δ = δ(R,X) – δ(S,X)= δ(R,X) – δ(R,–X) = δ(1st fr.) – δ(2nd fr.) 。
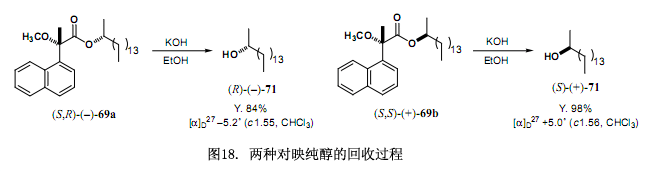
第二步是回收對映純醇與手性MαNP 酸3如圖18 例子所證實,分離后的酯經(jīng)溶劑分解作用生成對映純醇。回收的手性MαNP酸3還可以重復(fù)利用。
什么是回收醇的對映體純度?在我們看來所獲得兩種非對映酯都是對映純的,因為如果我們使用的MαNP酸3是對映純的,他們能很好的用高效液相色譜分離開。MαNP酸3能夠用天然(–)-薄荷醇對映拆分,用手性固定相的氣相色譜測定,其對映體純度肯定是100%。
如上所述,即使不考慮分子結(jié)構(gòu)的簡單性,MαNP酸也具有優(yōu)越的對映拆分能力,在磁各向異性效應(yīng)方面手性酸3要優(yōu)于Mosher’s MTPA 酸與Trost’s MPA 酸,因而需要進(jìn)一步的討論。
11.通過氫質(zhì)子核磁共振或者質(zhì)譜法的新非對應(yīng)異構(gòu)體法來測定對映體過量43)
怎樣通過不對稱合成或者酶促反應(yīng)確定所獲得的手性化合物的對映體過量(% ee)?因為手性分子化學(xué)在有機合成中的重要性持續(xù)增加,因而急需要新型的、準(zhǔn)確的方法來測定對映體過量。對映體過量(% ee)有如下定義:
% ee = 100{ | (R) – (S) | }/{(R) + (S)} (1)
上面已經(jīng)說到我們開發(fā)了一種新的手性磁各向異性試劑MαNP 酸3(圖19),它是一種通過氫質(zhì)子核磁共振譜圖來測定手性醇絕對構(gòu)型的較強功能試劑38-41)。手性酸3的另一個特點就是它具有很強的對映拆分外消旋醇的能力,特別是通過高效液相色譜對映分解無環(huán)脂肪醇,諸如MαNP酯類。例如經(jīng)MαNP酸 (S)-(+)-3酯化得到的消旋2-十六醇(±)-71,用硅膠高效液相色譜(正己烷/乙酸乙酯 = 20:1;分離因數(shù)α = 1.93 和分離度 Rs = 3.68)很容易分離所獲得的非對映體混合物。經(jīng)過酯的溶劑分解作用很容易得到2-十六醇-71的兩個純的對映體,這兩個對映體分別作為第一、第二洗脫組分被洗脫出來。MαNP 酸3的這些優(yōu)良特性有助于我們利用氫質(zhì)子核磁共振和質(zhì)譜圖法來開發(fā)新的非對映體法測定對映體過量43)。

有很多種測定手性化合物對映體過量的方法:1)對比對映體化合物的比旋光度[α]D值或圓二色譜強度;2)采用手性固定相的高效液相色譜或氣相色譜進(jìn)行對映分離44);3)利用手性有機金屬位移試劑的氫質(zhì)子核磁共振檢測45);4)通過高效液相色譜或氫質(zhì)子核磁共振檢測來分離由手性衍生劑所合成的非對映體46);5)通過利用手性衍生劑或手性主-客體絡(luò)合物做質(zhì)譜檢測47-50);使用對映分解動力學(xué)方案的方法51)。以上這些方法都具有各自的優(yōu)缺點,例如在一些情況下通過使用標(biāo)準(zhǔn)樣品的已知對映體過量值來確定對映體化合物的數(shù)據(jù)和校準(zhǔn)曲線;在另外一些情況下峰變寬就擾亂了峰值強度的精確測定。
對于使用手性衍生劑的非對映體方法,總會伴隨著動力學(xué)拆分的影響,因此最重要的問題是如何評價這種方法。如果衍生化反應(yīng)獲得了100%產(chǎn)率,那么就可以排除動力學(xué)拆分影響;但是,反應(yīng)要達(dá)到100%的產(chǎn)率是不切實際的。另外,基于動力學(xué)分解效應(yīng)的方法包括近似方法和需要校準(zhǔn)曲線;在個別情況下依據(jù)以下粗略近似來測定對映體過量。手性衍生劑的兩個對映體分別與手性底物反應(yīng),我們檢測了所生成的產(chǎn)物的產(chǎn)率,當(dāng)它們各自反應(yīng)的產(chǎn)率相似時,可以簡單地認(rèn)為動力學(xué)拆分影響可以忽略不計,不用對動力學(xué)拆分效應(yīng)進(jìn)行校正就能直接計算對映體過量的值。用非對映方法測定對映體過量時經(jīng)常受動力學(xué)拆分效應(yīng)干擾,不過就像下面所說,在徹底消除動力學(xué)拆分效應(yīng)的情況下我們依據(jù)氫質(zhì)子核磁共振與質(zhì)譜已經(jīng)成功的率先發(fā)展一種新型的對映體法來測定對映體過量43)。我們研發(fā)的方法優(yōu)點之一是使用質(zhì)譜保證了微量成分的高靈敏度檢測。
12.測定對映體過量新型方法的原則和步驟:徹底消除動力學(xué)拆分效應(yīng)
利用2-十六醇71作為試驗樣品來解釋測定對映體過量新型方法的原則和步驟,如圖20所示一份大約1:1的(S)-(+)-3MαNP 酸和氘標(biāo)記的(R)-(–)-3-d3 MαNP酸混合物與手性醇71反應(yīng)(0-100%的對映體過量)生成酯的非對映體混合物(圖 20, R = H, n = 3)。
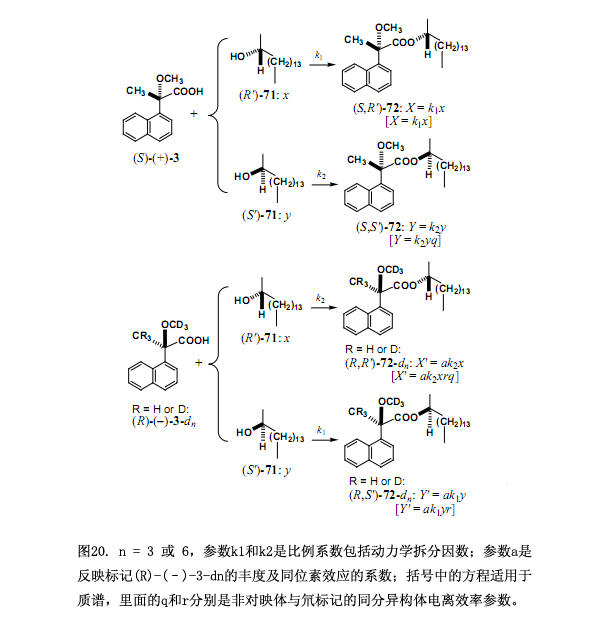
(R')-71 和(S')-71的組成分別定為x和y,其中x + y = 1,同樣的所生成的酯的組分被定為:酯 (S,R')-72, X;(S,S')-72, Y; (R,R')-72-d3, X';(R,S')-72-d3, Y'。酯的總量用以下公式表示:X = k1x 和 Y = k2y,在這里k1、k2是比例系數(shù)包括動力學(xué)拆分因數(shù)(注意這些系數(shù)都不是速率常數(shù)),由于酯(S,S')-72 和(R,R')-72-d3相互是對映體,相同的系數(shù)k2用來定義(R,R')-72-d3的量:X' = ak2x,此處a是反映形成酯的(R)-(–)-3-d3豐度與同位素效應(yīng)的系數(shù);對于另一個酯(R,S')-72-d3, Y' = ak1y。采用比例X/Y',這些方程簡化成方程(1),方程(1)中消去了比例系數(shù)也包括動力學(xué)拆分因數(shù)的k1。
X/Y' = (k1x)/(ak1y) = (1/a)x/y (2)
同樣的k2也被消去了
X'/Y = (ak2x)/(k2y) = (a)x/y (3)
由方程(2)與方程(3)作用得到方程(4),這里反映(R)-(–)-3-d3豐度和同位素效應(yīng)的系數(shù)a也被消去了。結(jié)果等于醇組分比(x/y)的平方。
(X/Y')(X'/Y) = [(1/a)x/y][(a)x/y] = (x/y)2
= [(1st,M)/(1st,M+3)][(2nd,M+3)/(2nd,M)] (4)
利用硅膠高效液相色譜(22φ × 300 mm,正己烷/乙酸乙酯 = 20:1)分離這些非對映體酯的混合物(圖21)。第一部分包含酯(S,R')-72 和(R,S')-72-d3,可以從MαNP部分的甲氧基與甲基官能團(tuán)的氫質(zhì)子核磁共振峰強度來確定各組分比例(X/Y') = [(1st,M)/(1st,M+3)],也即是甲氧基的峰強度對應(yīng)X,而甲基的峰強度對應(yīng)X+Y';相似的處理也適用于含有(S,S')-72 和(R,R')-72-d3的組分2,由氫質(zhì)子核磁共振峰強度得到比值(X'/Y) = [(2nd,M+3)/(2nd,M)],將上面的比值帶入到方程(4)利用x+y = 1的關(guān)系中計算(x/y)2,因而就確定了醇71的對映體過量(% ee)。
13.氫質(zhì)子核磁共振測定對映體過量應(yīng)用(% ee)
甲基-2-羥基-2-(1-萘基)丙酸跟CD3I(氘原子組分>99.5 %)經(jīng)甲基化反應(yīng)生成氘標(biāo)記的(R)-(–)-3-d3MαNP 酸,隨后水解并經(jīng)(–)-薄荷醇對映拆分。(S)-(+)-3 和(R)-(–)-3-d3混合物(比, 1:0.987, 總量8.1 mg, 0.0349 mmol)可以與測試樣品手性2 -十六醇(71, 9.237 mg, 1.09 × 0.0349 mmol, 60.9% 對映體過量按重量計算)反應(yīng),生成酯72的非對映體混合物,該混合物再由硅膠高效液相色譜(正己烷/乙酸乙酯 20:1)分離(圖21)。
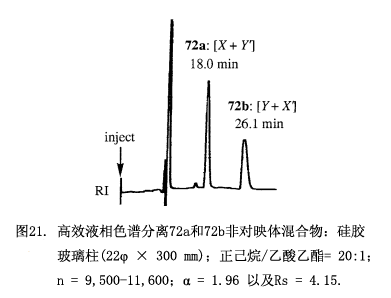
從第一組分的氫質(zhì)子核磁共振圖譜可以確定酯(S,R')-72和 (R,S')-72-d3的組成:X = 1.00, Y' = 0.22, X/Y' = 4.54,類似的從第二組分的氫質(zhì)子核磁共振圖譜確定(S,S')-72 和 (R,R')-72-d3 的組成:Y = 1.00, X' = 3.60, X'/Y = 3.60。結(jié)果為(X/Y')(X'/Y) = 16.3636,x/y = 4.045,再根據(jù)x+y = 1得出x = 0.8017 及 y = 0.1982,并獲得60.4%對映體過量,這個值與按重量計算的對映體過量60.9%相符合,本方法已經(jīng)用于10種71測試樣品,其對映體過量從0-90%。如圖22所示由氫質(zhì)子核磁共振所獲得的對映體過量與按重量計算的值相一致,平均誤差為±0.4% 對映體過量,而最大誤差1.3% 對映體過量。這些結(jié)果清楚的驗證了我們測定對映體過量方法的方案和原理。當(dāng)我們將5種測試樣品的對映體過量擴(kuò)大到92-100%時,其偏差變大:平均誤差±1.2% 對映體過量,最大誤差6.3%對映體過量。如此大的偏差可能是由于這些檢測僅限于測試氫質(zhì)子核磁共振弱峰的強度。為了克服這個問題我們采用更為靈敏的質(zhì)譜來測定組成。

14.質(zhì)譜確定對映體過量及應(yīng)用
就質(zhì)譜而言,非對映體(S,R')-72和(S,S')-72的電離效率不盡相同,氘取代酯(R,S')-72-dn與非取代酯(S,R')-72也不同。因此各異構(gòu)體電離效率因數(shù)(f)定義如下:酯(S,R')-72,f = 1,X = k1x;酯(S,S')-72,f = q 以及Y = k2yq;(R,S')-72-dn (n = 3 或 6), f = r 與Y' = ak1yr;(R,R')-72-dn (n = 3或6), f = rq 與 X' = ak2xrq。在這里由于非對映體結(jié)構(gòu)的差別q是相對電離效率,r跟氘取代有關(guān);做比值X/Y',方程(1)就變成:
X/Y' = (k1x)/(ak1yr) = (1/ar)x/y (2')
類似的方程(2)變形為
X'/Y = (ak2xrq)/(k2yq) = (ar)x/y (3')
因此,方程(3)變?yōu)?/p>
(X/Y')(X'/Y) = [(1/ar)x/y][(ar)x/y] = (x/y)2
= [(1st,M)/(1st,M+n)][(2nd,M+n)/(2nd,M)] (4')
由于所有的電離效率系數(shù)都被取消掉,測定對映體過量同樣的方案適用于質(zhì)譜測定。
采用質(zhì)譜測定對映體過量的方法基本適用于三氘取代的(R)-(–)-3-d3MαNP酸,在質(zhì)譜中3個m/z單位(M+3 vs. M)不足以避免自然豐度同位素峰的重疊,因此我們需要做一系列的修正,在修正背景后,跟同位素峰重疊,氘的含量,外消旋體(0%對映體過量),在(0-100% 對映體過量)范圍內(nèi)都很好的符合:平均誤差為±0.7%對映體過量;最大誤差是1.9%對映體過量。然而,這些修正針對實際應(yīng)用就太復(fù)雜了,因此我們選擇了六氘取代的(R)-(–)-3-d6MαNP 酸。
2-(1-萘基)-2-乙醛酸乙酯和CD3I (氘原子含量 >99.5 %)經(jīng)Grignard反應(yīng)制備了對映體手性酸(R)-(–)-3-d6,然后用CD3I (氘原子含量>99.5 %)對其甲基化反應(yīng),再經(jīng)水解、用(–)-薄荷醇對映拆分。對于此手性酸(R)-(–)-3-d6,由于6個m/z單位(M+6 vs.M)的差別,自然豐度的同位素峰重疊完全可以忽略不計。

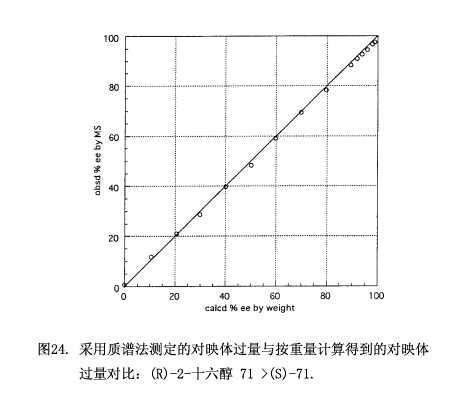
接下來使用測試樣品71來檢查這MS光譜法(按重量計算69.77%對映體過量),由(S)-(+)-3和(R)-(–)-3-d6(二者比:1:0.994)制備非對映體酯混合物,該混合物經(jīng)硅膠高效液相色譜(正己烷/乙酸乙酯 20:1)分離。從第一組分的質(zhì)譜圖23(a)可以看出,用背景校正法測得酯(S,R')-72 (M+ m/z 454) 和酯 (R,S')-72-d6 (M+ m/z 460)的組成:X = 52.284, Y' = 11.221, X/Y' = 4.6594;類似的從第二組分的質(zhì)譜圖23(b)來看(S,S')-72 (M+ m/z 454)和(R,R')-72-d6 (M+ m/z 460)的組成:Y = 8.607,X' = 55.278, X'/Y = 6.4224。結(jié)果是(X/Y')(X'/Y) = 29.9245, x/y = 5.4703. 由于 x+y = 1,所以得出x = 0.845448及y = 0.154552,還得到69.09%對映體過量,此對映體過量跟用按重量法計算的對映體過量69.77%相吻合。
質(zhì)譜法已經(jīng)用于測定如圖24所示的15種對映體過量0-100%的測試樣品71,由質(zhì)譜法所得到的對映體過量值跟按重量計算的對映體過量值相一致,平均誤差是±1.08%對映體過量,最大誤差為1.79%對映體過量;與氫質(zhì)子核磁共振圖譜不同的地方就是,即使在90-100%對映體過量范圍內(nèi)數(shù)據(jù)點也呈直線關(guān)系。質(zhì)譜法要比氫質(zhì)子核磁共振法靈敏,因此適用于測定高對映體過量的樣品。盡管我們做的樣品(R)-2-十六醇 71 > (S)-71,但是按同樣的方案也可以得出樣品(S)-2-71 > (R)-71。
因此,在徹底消除動力學(xué)拆分效應(yīng)情況下我們成功的開發(fā)了新的非對映體法來測定對映體過量43),這種方法中值得強調(diào)的是其既不需要對映純化合物數(shù)據(jù)也不需要制備使用已知對映體過量樣品校準(zhǔn)曲線,還不需要動力學(xué)拆分效應(yīng)數(shù)據(jù)。該法的優(yōu)越性就是以采用高效液相色譜分離非對映體為代價所得到。此方法還需在精確度上進(jìn)一步改進(jìn),現(xiàn)在正在調(diào)查研究除了醇以外還能否用于其他的化合物,Chemistry and Biology (Kagaku to Seibutsu)52)文獻(xiàn)已經(jīng)報道了這部分的詳細(xì)描述。
15.結(jié)論
我們研發(fā)了幾種手性助劑,尤其是手性羧酸。利用非對映高效液相色譜法成功的運用這些手性衍生劑對映拆分了醇,通過X-射線晶體學(xué)或者氫質(zhì)子核磁共振各向異性法測定了其絕對構(gòu)型,根據(jù)氫質(zhì)子核磁共振與質(zhì)譜法測定了對映體過量。X-射線晶體學(xué)法使用一個內(nèi)參比物是測定絕對構(gòu)型的最佳方法,但是很難得到理想單晶。在這種情況下,使用MαNP酸的氫質(zhì)子核磁共振法,該酸無需結(jié)晶過程就能的有效的測定絕對構(gòu)型。在對映拆分中手性二氯代鄰苯二甲酸與MαNP酸二者互補,當(dāng)用一種手性衍生劑拆分失敗后,就考慮用另外一個酸。上述方法在實驗室制備100%對映體純度對映純醇功能甚強,并能同時測定其絕對構(gòu)型。進(jìn)一步說,我們的方法適用于各種化合物,對映體的大量合成以及現(xiàn)在所進(jìn)行的提升質(zhì)譜測定對映體過量的質(zhì)量。
參考文獻(xiàn)
1) Bijvoet, J. M.; Peerdeman, A. F.; Van Bommel, A. J. Nature 1951, 168, 271.
2) Harada, N.; Nakanishi, K. Circular Dichroic Spectroscopy-Exciton Coupling in Organic Stereochemistry; University Science Books: Mill Valley, Calif., and Oxford University Press: Oxford, 1983.
3) Harada, N.; Soutome, T.; Murai, S.; Uda, H. Tetrahedron: Asymmetry 1993, 4, 1755.
4) Harada, N.; Soutome, T.; Nehira, T.; Uda, H.; Oi, S.; Okamura, A.; Miyano, S. J. Am. Chem. Soc. 1993, 115, 7547 .
5) N.; Hattori, T.; Suzuki, T.; Okamura, A.; Ono, H.; Miyano, S.; Uda, H. Tetrahedron: Asymmetry 1993, 4, 1789.
6) Hattori, T.; Harada, N.; Oi, S.; Abe, H.; Miyano, S. Tetrahedron: Asymmetry 1995, 6, 1043.
7) Toda, F. Top. Curr. Chem. 1987, 140, 43. Toda, F. Inclusion Compounds; Atwood, J. L.; Davis, J. E.; MacNicol, D. D., Ed.; Oxford University Press: Oxford, 1991; Vol. 4, 126-187. Toda, F. Advances in Supramolecular Chemistry; Gokel, G. W., Ed.; JAI Press: London, 1992; Vol. 2, 141-191.
8) Toda, F.; Tanaka, K.;Miyahara, I.; Akutsu, S.; Hirotsu, K. J. Chem. Soc., Chem. Commun. 1994, 1795. Toda, F.; Tanaka, K.; Leung, C. W.; Meetsma, A.; Feringa, B. L. J. Chem. Soc., Chem. Commun. 1994, 2371. Toda, F.; Tanaka, K.; Watanabe, M.; Abe, T.; Harada, N. Tetrahedron: Asymmetry 1995, 6, 1495-1498.
9) Toda, F. Yuki Gosei Kagaku Kyokaishi (J. Synth. Org. Chem. Jpn.) 1990, 47, 1118.
10) Ohtani, I.; Kusumi, T.; Kashman, Y.; Kakisawa, H. J. Am. Chem. Soc. 1991, 113, 4092. Kusumi, T.; Takahashi, H.; Ping, X.; Fukushima, T.; Asakawa, Y.; Hashimoto, T.; Kan, Y.; Inouye, Y. Tetrahedron Lett. 1994, 35, 4397. Kusumi, T.; Takahashi, H.; Hashimoto, T.; Kan, Y.; Asakawa,Y. Chem. Lett. 1994, 1093.
11) Seco, J. M.; Latypov, Sh. K.; Quinoa, E.; Riguera, R. Tetrahedron Lett. 1994, 35, 2921. Latypov, Sh. K.; Seco, J. M.; Quinoa, E.; Riguera, R. J. Org. Chem. 1995, 60, 504. Seco, J. M.; Latypov, Sh. K.; Quinoa, E.; Riguera, R. Tetrahedron 1997, 53, 8541.
12) Trost, B. M.; Belletire, J. L.; Godleski, S.; McDougal, P. G.; Balkovec, J. M.; Baldwin, J. J.; Christy, M. E.; Ponticello, G. S.; Varga, S. L.; Springer, J. P. J. Org. Chem. 1986, 51, 2370.
13) Orui, H. Yuki Gosei Kagaku Kyokaishi (J. Synth. Org. Chem. Jpn.) 1998, 56, 591. Fukushi, K. Nippon Nogeikagaku Kaishi 1998, 72, 1345.
14) Review, J. Chromatogr. A 2001, 906, 1-482. Yamamoto, C.; Okamoto, Y. Maku 2000, 25, 277. Kobayashi, Y.; Matsuyama, A.; Onishi, A. Fine Chemicals 2000, 29, 59.
15) Stereoselective Biocatalysis; Patel, R. N. Ed.; Marcel Dekker: New York, 2000.
16) Harada, N.; Ochiai, N.; Takada, K.; Uda, H. J. Chem. Soc., Chem. Commun. 1977, 495.
17) Harada, N.; Ono, H.; Nishiwaki, T.; Uda, H. J. Chem. Soc., Chem. Commun. 1991, 1753.
18) Weismiller, M. C.; Towson, J. C.; Davis, F. A. Organic Syntheses 1990, 69, 154.
19) Nehira, T.; Harada, N. to be published.
20) Toyota, S.; Miyasaka, T.; Matsumoto, Y.; Matsuo, T.; Oki, M. Bull. Chem. Soc. Jpn. 1994, 67, 1680.
21) Toyota, S.; Akinaga, T.; Kojima, H.; Aki, M.; Oki, M. J. Am. Chem. Soc. 1996, 118, 11460.
22) Toyota, S. Enantiomer 1999, 4, 25.
23) Harada, N.; Nehira, T.; Soutome, T.; Hiyoshi, N.; Kido, F. Enantiomer 1996, 1, 35 .
24) Harada, N.; Koumura, N.; Robillard, M. Enantiomer 1997, 2, 303.
25) Harada, N.; Koumura, N.; Feringa, B. L. J. Am. Chem. Soc. 1997, 119, 7256.
26) Toyota, S.; Yasutomi, A.; Kojima, H.; Igarashi, Y.; Asakura, M.; Oki, M. Bull. Chem. Soc. Jpn. 1998, 71, 2715.
27) Harada, N.; Vassilev, V. P.; Hiyoshi, N. Enantiomer 1997, 2, 123.
28) Harada, N.; Hiyoshi, N.; Vassilev, V. P.; Hayashi, T. Chirality 1997, 9, 623.
29) Harada, N.; Fujita, K.; Watanabe, M. Enantiomer 1997, 2, 359.
30) Fujita, K.; Harada, N. to be published.
31) Koumura, N.; Harada, N. to be published.
32) Harada, N.; Fujita, K.; Watanabe, M. Enantiomer 1998, 3, 64.
33) Harada, N.; Fujita, K.; Watanabe, M. J. Phys. Org. Chem. 2000, 13, 422.
34) Kuwahara, S.; Watanabe, M.; Harada, N.; Koizumi, M.; Ohkuma, T. Enantiomer 2000, 5, 109.
35) Watanabe, M.; Kuwahara, S.; Harada, N.; Koizumi, M.; Ohkuma, T. Tetrahedron: Asymmetry 1999, 10, 2075.
36) Taji, H.; Harada, N. to be published.
37) Kosaka, M.; Watanabe, M.; Harada, N. Chirality 2000, 12, 362.
38) Kuwahara, S.; Fujita, K.; Watanabe, M.; Harada, N.; Ishida, T. Enantiomer 1999, 4, 141.
39) Ichikawa, A.; Hiradate, S.; Sugio, A.; Kuwahara, S.; Watanabe, M.; Harada, N. Tetrahedron: Asymmetry 1999, 10, 4075.
40) Harada, N.; Watanabe, M.; Kuwahara, S.; Sugio, A.; Kasai, Y.; Ichikawa, A. Tetrahedron: Asymmetry 2000, 11, 1249.
41) Taji, H.; Kasai, Y.; Sugio, A.; Kuwahara, S.; Watanabe, M.; Harada, N.; Ichikawa, A. Chirality 2002, 14, 81.
42) Harada, N.; Watanabe, M.; Kuwahara, S.; Kosaka, M. Yuki Gosei Kagaku Kyokaishi (J. Synth. Org. Chem. Jpn.) 2001, 59, 985.
43) Taji, H.; Watanabe, M.; Harada, N.; Naoki, N.; Ueda, Y. Org. Lett. 2002, 4, 2699.
44) Okamoto, Y.; Hatano, K.; Aburatani, R.; Hatada, K. Chem. Lett. 1989, 715.
45) Kabuto, K.; Sasaki, Y. J. Chem. Soc., Chem. Commun. 1987, 670.
46) Dale, J. A.; Mosher, H. S. J. Am. Chem. Soc. 1973, 95, 512.
47) Sawada, M.; Yamaoka, H.; Takai, Y.; Kawai, Y.; Yamada, H.; Azuma, T.; Fujioka, T.; Tanaka, T. Int. J. Mass Spectrom. 1999, 193, 123. Sawada, M.; Takai, Y.; Iwamura, H.; Yamada, H.; Takahashi, S.; Yamaoka, H.; Hirose, K.; Tobe, Y.; Tanaka, J. Eur. J. Mass Spectrom. 2001, 7, 447.
48) Guo, J.; Wu, J.; Siuzdak, G.; Finn, M. G. Angew. Chem. Int. Ed. 1999, 38, 1755 .
49) Grigorean, G.; Ramirez, J.; Ahn, R. H.; Lebrilla, C. B. Anal. Chem. 2000, 72, 4275.
50) Yao, Z.-P.; Wan, T. S.; Kwong, K.-P.; Che, C.-T. Anal. C
統(tǒng)計數(shù)據(jù)
共收錄化學(xué)品數(shù)據(jù)
147579 條
公告
更多>
-
微信掃一掃
關(guān)注:物競化學(xué)品數(shù)據(jù)庫
-
物竟數(shù)據(jù)庫 手機版
國內(nèi)首款化工類專業(yè)手機應(yīng)用
-
微博賬號
wjhxp
快速導(dǎo)航
化學(xué)品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
關(guān)于物競
物競數(shù)據(jù)庫是一個全面、專業(yè)、專注,并且免費的中文化學(xué)品信息庫,為學(xué)生、學(xué)者、化學(xué)品研究機構(gòu)、檢測機構(gòu)、化學(xué)品工作者提供專業(yè)的化學(xué)品平臺進(jìn)行交流。
數(shù)據(jù)庫采用全中文化服務(wù),完全突破了中英文在化學(xué)物質(zhì)命名、化學(xué)品俗名、學(xué)名等方面的差異,所提供的數(shù)據(jù)全部中文化,更方便國內(nèi)從事化學(xué)、化工、材料、生物、環(huán)境等化學(xué)相關(guān)行業(yè)的工作人員查詢使用。
關(guān)注我們
-
微信賬號:物競化學(xué)品數(shù)據(jù)庫

-
微博賬號:wjhxp
聯(lián)系我們
上海市延長路149號上海大學(xué)科技園412室
公司總機: 021-56389801
訂購電話: 4007001514
傳真電話: 021-56389802
客服電話: 021-56332350
電子郵件: wingch@basechem.org
Copyright ? 物競數(shù)據(jù)庫 2009-2026.All Rights Reserved. 關(guān)于數(shù)據(jù)庫 | 關(guān)于物競 | 法律聲明 | 熱門關(guān)鍵字
 滬公網(wǎng)安備 31010602001115號
滬公網(wǎng)安備 31010602001115號