具有生物活性的單氟化合物的合成
- 2012-05-25
- 專題
將氟引入一生物活性化合物和一農藥的某一位置,可顯著減少化合物的毒性,或改善藥效。這可能是由模仿和阻斷作用造成的。模擬作用是有幾天的代謝途徑很容易納入帶有C-F鍵化合物的現象。這是因為氟原子很小,所以C-H鍵和C-F鍵之間沒有明顯的立體差異。阻斷作用是氟原子的電負性降低周圍電子密度的現象,并阻擋程序中的某些反應,例如氧化反應。氟類固醇和5-氟-尿嘧啶衍生物是氟化作用的例子(圖1)。
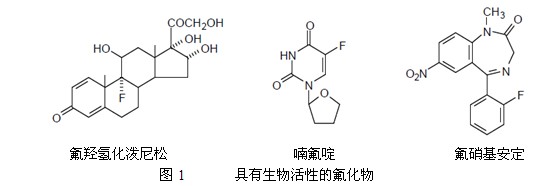
由于很少出現天然的含氟化合物,所以必須在合成的某一階段使有機化合物與氟素化合。雖然把氟氣和氟化氫作為氟源,但是它們的毒性和腐蝕性很大,而且需要特殊的儀器和技術來操作它們。因此,已經發明出代替氟化劑的試劑,在實驗室中使用它很容易在化合物選定位置上引入氟原子。粗略的將氟化劑劃分為兩類:親核額和親電的。親核氟化劑是這些作為反應活性物種的氟化物陰離子。親電氟化劑是這些作為反應活性物種的缺電子氟化物。另外,作為氟源使用含氟的構建模塊,它在一個分子中同時擁有氟原子和可替換的功能團。
1.親核氟化試劑
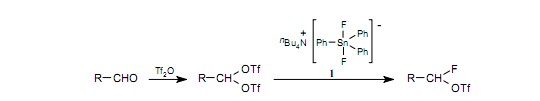
最基礎的親核氟化試劑是氟化氫,氟化合物的工業產品大量使用它。然而,由于它的毒性、腐蝕性以及由H-F的高鍵能造成的低反應活性,在實驗室中極少使用氟化氫。像KF、CsFB和Bu4N·F這樣的親核氟化試劑很容易得到。這些氟化試劑易潮,而不幸的是在這些成分中的水分與氟化物陰離子形成強氫鍵,并導致反應活性的降低。有人已經研制出了“裸”氟化物陰離子,即氟化物陰離子完全自由于氫鍵。舉個例子,三丁基胺二氟三丙基錫酸鹽1(由M.Gingrasas報道作為Bu4N·F的替代試劑)是防潮的,在高溫中有很好的穩定性而且在有機溶劑中有很高的溶解度。1)A.G.Martines等用物質1從醛合成gem-氟三氟。2)
作為一種強氟化試劑使用TASF2。3)它易潮,但很容易獲得無水晶體。三(二甲基氨基)锍的電荷并不是集中在S原子上,而是擴散到整個基團。因此,(Me2N)3S+ 和Me3SiF2-間的靜電作用比較弱。Me3SiF2-分離成Me3SiF和F-,釋放了一個氟化物陰離子源后在有機溶液中是易溶的。W.A.Szarek等已經演示了在溫和環境下,來自糖三氟的二氧氟糖的合成方法。

在很多種合成工藝中使用DAST3,用它使氟原子立體具體取代羥基團。4)A.P.Kozikowski等已經發明了兩步從白堅木醇生產(-)-氟-myo-纖維醇的方法。5)他們稱,在這種方法中,DAST3與白堅木醇兩個軸向羥基中的一個反應生成一個DAST中間體,它可以極大地增強羥基的離去能力。與硬酯反演協同,通過氟化物陰離子作用,使一個容易的羥基親核取代發生成為可能。物質3高溫下不穩定,而且經過加熱大幅度分解。然而,通常在室溫或較低溫度下,在一個短時間完成使用3的反應。

促進氟化物陰離子攻擊的另一種方法是通過產生碳正離子。通過氧化有機化合物可以生產碳正離子。因此,氧化劑和氟化試劑的聯合使用是簡單的氟化作用成為可能。另外,通過選擇一種氧化劑,選擇性氟化成為可能。使用三丁基氨基二氫三氟化物4和NIS,Kuroboshi等已經獲得從烯烴到氟碘代烷烴的合成方法。6)

電解氧化也可作為一種氧化有機化合物的方法。因為沒有使用氧化劑的簡單反應系統,所以通過這種方法可以很容易對目標物質進行提純。
2. 親電氟化試劑
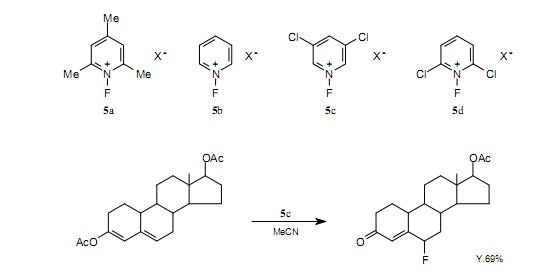
在親電氟化試劑中,基本試劑是氟氣,由于它的較高反應活性所以并不適用于部分氟化,同時它有強毒性。Umerzawa等報道說,N-氟-吡啶鹽5是一種穩定并且易于操作的晶體,而且它具有較高的活性。7)通過將吸電子基團或給電子基團引入吡啶環中,可以增加或減小氟化能力。舉個例子,氟化能力以5a,5b,5c和5d的順序增加,下面顯示了它們的結構。通過為實驗選擇一個合適的反映試劑,抑制副反應和改善收率是可能的。
在1988年,E.Differding等第一次成功合成非對稱氟化試劑,N-氟樟腦磺內酰胺6。8)這允許以70%的對映體過量的碳負離子非對稱氟化。

3. 含氟構建模塊
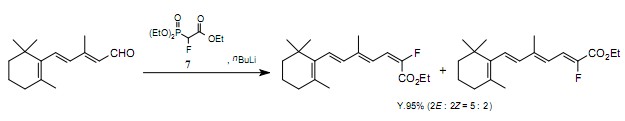
最近,同時擁有一個氟原子和可替代功能團的含氟構建模塊,作為一種易得的氟源使用。特別是,在同一碳原子上同時含有一個氟原子和一個吸電子基團的化合物,作為一種對合成生理活性化合物有效地含氟構建模塊來使用。為生成能夠與親電子試劑反應的氟-碳負離子,用堿與這類化合物進行處理。舉個例子,在堿存在時,三乙基2-氟-2-磷酰基乙酸鹽79)生成一個氟-碳負離子。與羰基化合物一起,陰離子的Hormer-Emmons反應生成α-氟-α,β-不飽和酸酯。這些酯能被還原成醛和醇,而且它們是生成很多生理活性化合物的非常有用的前驅體。合成的化合物(維他命A衍生物10),昆蟲外生物激素11)和有殺蟲作用的擬除蟲菊酯12))是與物質7一起生成,具有生理活性含氟物質的主要實例。
O.Piva觀察到通過254nm光的照射,在堿存在時,α-氟-α,β-不飽和酸酯可轉化成α-氟-β,γ-不飽和酸酯。如果一種光學活性的胺作為堿來使用,可優先得到光學對映體中的任一種。13)
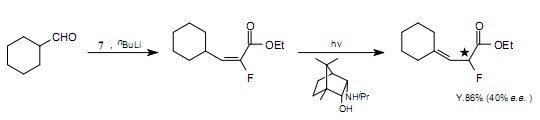
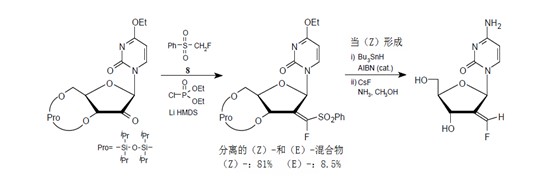
氟甲基苯基砜在同一個碳原子上具有氟原子和磺酰基,在堿存在時生成氟-碳負離子。14)這些陰離子與羰基化合物反應生成α-氟-β-羥基苯基砜衍生物。這種砜可轉化成α-氟-α,β-不飽和砜和氟乙烯基衍生物。J.R.McCarthy等報道了一種方法,這種方法使用物質8能夠將氟烯基單元立體選擇地引入核苷。15)
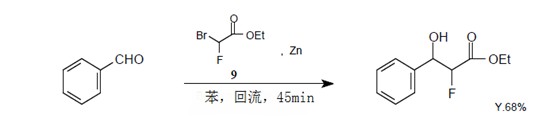
甲基溴氟乙酸9在同一個碳原子上具有一個溴原子,一個氟原子和一個羰基,與鋅反應生成瑞福馬斯基試劑。這種瑞福馬斯基試劑與羰基化合物反應生成α-氟-β-羥基酯,16)與N-(α-氨烷基)苯三唑化合物反應生成α-氟-β-氨基酯。17)Takeuchi等使用物質9合成了N-保護α-氟-α-氨基酯,而且報道了它的化學性質的詳細資料。18)
在這描述的氟化試劑和含氟構建模塊只需要普通的有機合成方法。生成含氟有機化合物不需要特殊儀器和技術。生成許多藥物和農藥已經使用了這些氟化試劑。
參考文獻
1) M. Gingras, Tetrahedron Lett., 32, 7381 (1991).
2) A. G. Martinez, J. O. Barcina, A. Z. Rys, L. R. Subramanian, Tetrahedron Lett., 33, 7787 (1992); idem, Synlett, 1993, 587.
3) W. A. Szarek, G. W. Hay, B. Doboszewski, J. Chem.Soc., Chem. Commun., 1985, 663; P. J. Card, W. D. Hitz, J. Am. Chem. Soc., 106, 5348 (1984).
4) L. N. Markovskij, V. E. Pashinnik, A. V. Kirsanov, Synthesis, 1973, 787; W. J. Middleton, J. Org. Chem., 40, 574 (1975); M. Hudlicky, Org. React., 35, 513 (1988); Wm. Rosenbrook, Jr., D. A. Riley, P. A. Lartey, Tetrahedron Lett., 26, 3 (1985).
5) P. A. Kozikowski, A. H. Fauq, J. M. Rusnak, Tetrahedron Lett., 30, 3365 (1989).
6) M. Kuroboshi, T. Hiyama, Tetrahedron Lett., 32, 1215 (1991).
7) T. Umemoto, K. Tomita, Tetrahedron Lett., 27, 3271 (1986).
8) E. Differding, R. W. Lang, Tetrahedron Lett., 29, 6087 (1988).
9) S. Kikuchi, T. Onosawa, Journal of Synthetic Organic Chemistry, Japan, 55, 88 (1997).
10) R. S. H. Liu, H. Matsumoto, A. E. Asato, M. Denny, Y. Shichida, T. Yoshizawa, F. W. Dahlquist, J. Am. Chem. Soc., 103, 7195 (1981); A. J. Lovey, B. A. Pawson, J. Med. Chem., 25, 71 (1982).
11) F. Camps, J. Coll, G. Fabrias, A. Guerrero, Tetrahedron, 40, 2871 (1984).
12) Ph. Coutrot, C. Grison, R. Sauvetre, J. Organoment. Chem., 332, 1 (1987).
13) O. Piva, Synlett, 1994, 729.
14) M. Inbasekaran, N. P. Peet, J. R. McCarthy, M. E. LeTourneau, J. Chem. Soc., Chem. Commun., 1985, 678.
15) J. R. MaCarthy, D. P. Matthews, D.M. Stemerick, E. W. Huber, P. Bey, B. J. Lippert, R. D. Snyder, P. S. Sunkara, J. Am. Chem. Soc., 113, 7439(1991).
16) E. T. McBee, O. R. Pierce, D. L. Christman, J. Am. Chem. Soc., 77, 1581(1955); S. Brandange, O. Dahlman, L. Morch, ibid., 103, 4452(1982).
17) A. R. Katriazky, D. A. Nichols, M. Qi, Tetrahedron Lett., 39, 7063(1998).
18) Y. Takeuchi, M. Nabetani, K. Takagi, T. Hagi, T. Koizumi, J. Chem. Soc. Perikin Trans. 1, 1991, 49; Y. Takeuchi, K. Takagi, T. Yamaba, M. Nabetani, T. Koizumi, J. Fluorine Chem., 68, 149(1994).
注:本文為提供者翻譯的,由于知識所限,其中錯誤在所難免,敬請原諒。如有問題可以查找原文。
快速導航
化學品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
關于物競
物競數據庫是一個全面、專業、專注,并且免費的中文化學品信息庫,為學生、學者、化學品研究機構、檢測機構、化學品工作者提供專業的化學品平臺進行交流。
數據庫采用全中文化服務,完全突破了中英文在化學物質命名、化學品俗名、學名等方面的差異,所提供的數據全部中文化,更方便國內從事化學、化工、材料、生物、環境等化學相關行業的工作人員查詢使用。
關注我們

-
微信賬號:物競化學品數據庫

-
微博賬號:wjhxp
聯系我們
上海市延長路149號上海大學科技園412室
公司總機: 021-56389801
訂購電話: 4007001514
傳真電話: 021-56389802
客服電話: 021-56332350
電子郵件: wingch@basechem.org
 滬公網安備 31010602001115號
滬公網安備 31010602001115號