氣體膨脹液體技術制備納米微粒研究進展
- 2012-05-21
- 專題
氣體膨脹液體(Gas Expanded Liquids, GXLs)是一類新的、用途廣泛的環(huán)境友好溶劑,它通常是由有機溶劑和高壓氣體組成的混合物[1,2]。GXLs是在超臨界流體(SCFs)基礎上發(fā)展起來的,常用的GXLs,通常其體積膨脹率可達5~10倍[3]。在CO2膨脹液體中,通過改變CO2的組成,可使膨脹液體從常規(guī)有機溶劑連續(xù)過渡到超臨界CO2(scCO2),CO2-GXLs的性質可通過改變操作壓力得以調節(jié);通常大量CO2的存在有利于傳質及氣體的溶解度,而極性有機溶劑的存在則可提高固體及液體溶質的溶解度。在壓力0~8MPa范圍內,氣體CO2可以溶解于許多醇、酮、醚及其酯類有機溶劑中[4],相對于SCFs而言,GXLs可在非常常溫和的壓力條件下有效地調節(jié)體系的極性、介電常數(shù)、溶解度等物理化學性質。在過程分離﹑微細顆粒沉積﹑促進聚合物處理以及作為催化反應的介質等方面,GXLs作為最理想溶劑已逐步得到認可。GXLs用作溶劑介質有如下4個優(yōu)勢:(1)較高的擴散系數(shù)和較低的粘度,減少了傳質的限制;(2)溶劑性質很容易由壓力來調節(jié);(3)下游產(chǎn)品處理和溶劑去除更為簡便;(4)提供了取代傳統(tǒng)有機溶劑的環(huán)境友好的介質。GXLs納米制備技術具有豐富的研究內容,是一個充滿活力的研究領域,是涉及綠色化學、化學熱力學、模擬計算和多種表征技術等相互交叉滲透的綜合利用技術。
納米材料研究是目前材料科學研究的一個熱點,納米技術也是21世紀最具有前途的科研領域。由于納米微粒所表現(xiàn)出的性質強烈地依賴于微粒的尺寸及形狀、微粒之間的距離和起保護作用的配體的性質[5],這就為我們期望利用GXLs 的溶劑化特性來實現(xiàn)控制納米微粒的尺寸及形狀、微粒之間的距離及介電環(huán)境,從而最終實現(xiàn)納米微粒的性質可控開辟了廣闊的空間。GXLs技術制備納米微粒是近幾年才發(fā)展起來的一項新技術,是制備微細材料技術的一種有效手段。本文簡要介紹了GXLs作為反應介質及過程處理溶劑的優(yōu)勢、GXLs超細材料制備技術及原理、GXLs技術在相關領域的應用及存在問題。
1 GXLs 作為反應介質及過程處理溶劑的優(yōu)勢
1.1 反應優(yōu)勢
由于許多化合物在SCFs,特別是scCO2中的溶解度較小,很大程度上限制了SCFs的應用。通過加入共溶劑,雖可改善化合物在scCO2中的溶解度,但其影響有限,而且1%~5%的共溶劑的加入很大程度上提高了混合體系的臨界壓力,不可避免地增加了工業(yè)過程的成本;相比之下,GXLs具有超強的溶解能力,它可在較低的壓力區(qū)間允許溶解高達10%~50%的有機溶劑,這也使其作為化學反應介質獨具優(yōu)勢[2,6~8]。與室溫有機溶劑相比,壓縮CO2的存在使GXLs具有較高的氣體混溶性,傳輸速率較之在常規(guī)有機溶劑或scCO2中可提高1到2個數(shù)量級。
1.2 過程優(yōu)勢
首先,較之在常規(guī)液體溶劑中可提高試劑氣體的溶解度,GXLs的操作壓力一般為幾個MPa ,與scCO2下通常幾十個MPa 的壓力相比,顯然更加溫和。其次,GXLs過程可使有機化合物(如簡單有機溶劑﹑離子液體[9]、油脂[10]﹑以及聚合物[11~17])的熔點降低,熔點的降低是由于溶融的有機化合物中溶解了部分氣體。此外,任何化學過程不可避免地牽涉到產(chǎn)物與反應物等的分離或產(chǎn)物的提純,其中分離或純化通常占到化學過程總成本的60%~80%,而GXLs 因具有常規(guī)液體無可比擬的流動性和超強的溶解力,從而使分離過程易于實現(xiàn),且分離成本大為降低。
2 GXLs 中超細材料制備技術﹑原理及應用
對納米材料制備技術的研究可進一步深化人們對材料本質、結構和特性的認識,也為新一代納米材料的設計與開發(fā)提供技術支持。利用GXLs 的相行為已經(jīng)開發(fā)了許多微粒制備方法,可以從無機﹑有機﹑有機金屬固體中制備微細材料(圖1)[18~20],其中有機微粒廣泛應用于顏料[21]﹑食品[22]﹑炸藥[23]﹑藥物化學[19,24,25]等領域。
GXLs-納米微粒制備原理是,GXLs技術通常利用氣體將溶液或純溶質進行膨脹,制備方法包括從溶液中沉淀微粒或先將溶質熔解然后通過凝固或冷凍的方式析出,微粒一旦形成,可借助GXLs 中的氣體達到干燥或粒徑窄化的目的。為防止納米微粒團聚,通常制備的微粒外面包覆有一配體保護層,由配體穩(wěn)定的納米顆粒在溶劑中的分散性是與溶劑的溶劑化強度密切相關的:在溶劑化較強的溶劑中,納米顆粒配體端與分散溶劑的相互作用較強,因而可以克服納米顆粒間的范德華引力而使納米顆粒穩(wěn)定地分散于溶劑中,當加入一定壓力的氣體如CO2后,由于CO2弱的溶劑化,使得整個混合溶劑的溶劑化能力減弱,導致混合溶劑與納米顆粒配體端的相互作用減弱,當混合溶劑的溶劑強度低于一定的閾值時,已經(jīng)分散的納米顆粒將會逐漸從GXLs 中沉淀析出,而粒徑最大的顆粒由于較強的范德華引力會首先自弱化的混合溶劑中沉淀析出。
目前,GXLs納米微粒制備方法主要有氣體飽和溶液微粒法(Particles from Gas-Saturated Solution, PGSS)﹑氣體抗溶劑法(Gas Antisolvent, GAS)﹑壓縮抗溶劑沉積法(Precipitation with Compressed Antisolvent,PCA)﹑氣溶膠溶劑萃取體系技術(Aerosol Solvent Extraction System, ASES)﹑超臨界流體強化溶液分散沉淀過程(Solution-Enhanced Dispersion by Supercritical Fluids, SEDS)﹑膨脹液體有機溶液減壓技術(Depressurization of an Expanded Liquid Organic Solution, DELOS)﹑反相乳液微粒沉淀技術(Precipitation of Particles from Reverse Emulsions)等,相應微粒制備過程圖例參見圖1。
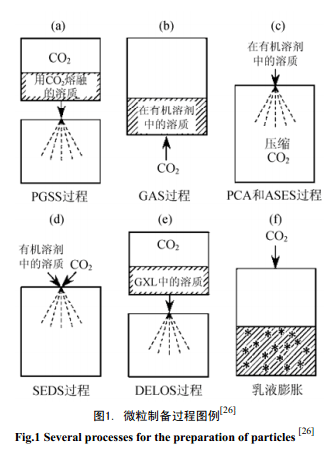
2.1 PGSS 過程
PGSS 的原理是利用氣體膨脹使得固體溶質溶解于GXLs 中,然后使溶有溶質的膨脹液體通過一個小的噴嘴,由于膨脹流體經(jīng)過噴嘴時產(chǎn)生的噴濺及排氣行為,致使該膨脹流體迅速降壓和冷卻,因此可獲得微細的粉末顆粒(圖1a),該方法只有在操作溫度接近目標化合物的熔點時才可行。Sencar-Bozic等[27]發(fā)現(xiàn),與未經(jīng)處理的材料相比,作為一種鈣離子通道模塊的鎳鐵雙蒎烯(其熔點范圍172~174℃),在185℃下經(jīng)PGSS過程所獲得的微粒不僅粒徑更小,而且在水中的溶解速度也更快,不足之處是該過程所需溫度太高,甚至還發(fā)現(xiàn)鎳鐵雙蒎烯在微粉化過程中會產(chǎn)生部分降解,因此這種方法對于許多藥品來說是不太實用的。為使PGSS過程可在低溫下可行,研究人員對由分子量為4000 的聚乙烯二醇和鎳鐵雙蒎烯組成的混合物的微粉化過程(操作溫度50℃)進行了評估,發(fā)現(xiàn)經(jīng)這種方法獲得的共沉淀微細粉末,未觀察到鎳鐵雙蒎烯的降解行為。利用類似原理,Shine等[28]介紹了一個制備聚已酸內酯膠囊型疫苗微粒的實例。
2.2 GAS 過程
GAS方法[29] 中CO2作為抗溶劑使用,利用CO2誘導溶液發(fā)生膨脹以觸發(fā)沉淀,膨脹液體中溶質的溶解度隨氣體壓力增大而逐漸降低,直至溶質沉淀析出。隨后,將容器內的物質轉出即可使固體與膨脹液體發(fā)生分離( 圖1b)。GAS技術的基礎是許多物質可溶解于有機溶劑,而不溶于高壓流體,同時,高壓流體在許多有機溶劑中溶解度很大,從而使這些有機溶劑發(fā)生體積膨脹,使其內聚能明顯降低,溶劑的溶解能力降低,導致溶質結晶析出。膨脹后的液體與常壓下的液體相比,具有更高的擴散系數(shù)和更低的粘度,GAS法制備的結晶中溶劑的含量比傳統(tǒng)法要少得多,大大提高了結晶的純度,而高壓流體起到抗溶劑作用。在抗溶劑過程中,析出產(chǎn)物的粒度和晶型可由體系的壓力、溫度以及高壓氣體的溶解速度等來控制。
2.3 PCA 及 ASES 過程
隨著GAS過程的不斷發(fā)展,出現(xiàn)了許多對GAS過程進行改進的抗溶劑技術,如壓縮抗溶劑沉積法(PCA)及氣溶膠溶劑萃取體系技術(ASES)[3]等,其中ASES 是在PCA基礎上改進的,同時PCA(圖1c)及ASES與GAS過程恰恰相反,是將溶液噴射到高壓的CO2 中,高壓CO2迅速溶于溶液滴,使之發(fā)生膨脹,降低了溶劑的溶解能力,極短時間內形成極高的過飽和度,使溶質快速析出,形成細微而均勻的顆粒。經(jīng)比較發(fā)現(xiàn),PCA過程獲得的微粒粒徑較之GAS方法更加細微。隨后,可利用新鮮的CO2將微粒表面的有機溶劑洗掉[18]。PCA方法還可用于制備含有CO2或其它壓縮氣體的特殊晶體[30,31]。而在PCA基礎上改進的ASES方法中,溶劑不僅發(fā)生膨脹,而且可完全溶于scCO2中,通過選擇CO2氣流方向與有機溶液的噴濺方向相同或逆向,可將有機溶劑從反應器中萃取并除去[32]。
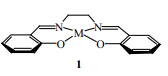
雖然大多數(shù)PCA或ASES過程更青睞有機化合物微粒的制備,但也有少數(shù)例外。例如作為活性催化劑,無定形的磷酸釩固體可用于將丁烷氧化成馬來酸酐,Hutchings等[33]通過磷酸和三氯氧釩的反應,將獲得的磷酸釩溶于異丙醇,接著利用PCA過程將磷酸釩從上述溶液中沉淀出來。Johnson等[34]首先制備出包含salen配體的金屬配合物(1),然后利用PCA技術從該配合物的二氯乙烷溶液中制備出長度達700 nm的棒狀微粒,該微粒作為多相催化劑或者在氣體捕獲方面具有潛在的應用價值。
2.4 SEDS 過程
SEDS過程[35]是將scCO2和含溶質的有機溶液經(jīng)一個同軸噴嘴同時充入反應器(圖1d),高速通過噴嘴的SCFs使溶液破碎成細小的液滴,同時將溶劑從該溶液中萃取出來,使析出的溶質顆粒得以快速干燥。SEDS過程中溶質和有機溶劑的接觸時間非常短暫,特別適用于難溶于SCFs而且對有機物比較敏感物料的微細顆粒的制備。SEDS可制備出較PCA或ASES過程尺寸更小的微粒。Subramaniam研究組改進的SEDS技術使用一種超聲噴嘴和scCO2作為驅動介質來形成藥物溶液的微型液滴,scCO2還可選擇性地從液滴中萃取溶劑,進而將藥物沉淀出來。利用SEDS方法已經(jīng)獲得了氫化可的松(hydrocortisone)﹑布洛芬(ibuprofen)﹑胰島素(insulin)及紫杉醇(paclitaxel)等藥品的納米級粉末[36~38] 。
2.5 DELOS 技術
在DELOS技術中,首先將溶質溶解于一種有機溶劑,接著充入氣體使溶液膨脹。因溶質的濃度足夠低,所以即使溶液發(fā)生體積膨脹也不會引起沉淀析出。但接下來的體系壓力驟降會導致已溶解的CO2迅速自溶液中逸出,氣體的迅速釋放引起的溫度劇烈降低足以誘導析出微細粉末狀的溶質(圖1e)。若在DELOS過程中使用1,1,1,2-四氟乙烷代替CO2,會有同樣的效果,但過程的操作壓力將會更低,從而DELOS更具優(yōu)勢[39]。
2.6 反相乳液微粒沉淀技術
利用GXLs還可以將溶解于反相乳液水相內的化合物以固體微粒形式沉積出來(圖1f),這項技術已經(jīng)用于蛋白質[40]及酶的提取[41]。例如,膨脹前的乳液由癸烷連續(xù)相(含表面活性劑)﹑水相(水/表面活性劑的摩爾比, w/o=20)及胰島素蛋白(trypsin protein)組成,在20℃時,將水相含有胰蛋白酶的反相乳液充入CO2 (操作壓力5 MPa)進行膨脹,即可自反相乳液中沉積出平均粒徑達10nm的胰蛋白酶微粒[40]。利用相關技術,也可將無機微粒沉淀析出[42,43]。比如,Zhang等[42]將兩種異辛烷-AOT-水的反相乳液混合,其中一種乳液其水核內含有ZnSO2,另一種則含有Na2S,將上述兩種乳液進行混合即可生成ZnS,最后通過CO2使乳液膨脹而收集得到ZnS微粒。
2.7 微粒過程處理(Particle Processing)
表面均勻﹑排列緊密的單層納米微粒薄膜具有許多潛在的應用價值,而常規(guī)薄膜制備方法是通過溶劑蒸發(fā)而從微粒的懸浮液中獲得理想的單層薄膜。該方法的弊端是,由于在去除溶劑時,存在著較高的界面張力及毛細作用力,會導致非常不均勻的微粒分布:一部分表面可能會富集目標微粒,而另一部分則可能缺乏目標產(chǎn)物。Roberts等[44,45] 向世人展示了GXLs制備納米薄膜的魅力:室溫下利用CO2使溶劑膨脹,然后升溫至40℃,同時伴隨新鮮的CO2進行多次清洗,最后即可成功地沉淀出分布均勻的單層膜,而且該方法中溶劑去除非常完全。Roberts等[46,47]還通過逐漸增加CO2的壓力來實施對納米微粒進行分級沉淀窄化的研究,他們已自GXLs 中成功制備了由烷基硫醇配體保護的金或銀的納米微粒。
Subramaniam等[36,48]借助PCA過程對微粒進行了表面鍍層。在PCA技術中,scCO2一方面驅使核心基底具有流動性,另一方面還可將溶劑從噴射到基底材料上的鍍層溶液中除去,最終將涂層沉積在基底表面上。例如,粒徑1~2mm 的粒狀玻璃及粒狀食用糖外表面通常涂敷一層薄薄的RG503H聚合物,該聚合物外殼既可做成微細顆粒型的,也可做成一個連續(xù)的微粒薄膜。與常規(guī)的空氣懸浮液Wurster涂膠過程相比,基于CO2的涂敷過程,進一步拓展了底物/溶劑組合范圍,使得在水溶性的底物表面涂敷溶質外殼成為可能。
3 GXLs 超細材料制備技術的優(yōu)勢、存在問題及研究趨勢
由于GXLs的溶劑強度的可設計性及獨特的傳輸特性,使得GXLs抗溶劑誘導沉淀在納微米顆粒制造方面具有非常重要的實用價值,在納米材料的合成、組裝及處理方面也蘊含著極大的潛能。近幾年來,有相關的報道了利用GXLs從AOT反相膠束中沉淀納米微粒的系列工作[43,49,50],Roberts等、Subramaniam等也已開展了涉及納米金屬顆粒[46,47]、納米金屬薄膜[44]、納米半導體微晶[51]以及對微粒進行表面鍍層[36,48]的系列研究。
3.1 環(huán)境及生產(chǎn)成本優(yōu)勢
傳統(tǒng)的基于有機溶劑的納米制備技術,其中大量揮發(fā)性有機溶劑作為抗溶劑使用,會造成較大的環(huán)境污染;而使用scCO2 的顆粒制備過程,相應的顆粒應用范圍較窄,且操作壓力和溫度也較高,尤其是,該方法還需使用較為昂貴的、在環(huán)境中難于降解的含氟化合物,因而對環(huán)境不友好。與傳統(tǒng)方法相比,GXLs綠色環(huán)保;與SCFs制備方法相比,GXLs過程易于實現(xiàn),操作條件更加溫和,制備成本大為降低。
3.2 技術優(yōu)勢
以核殼半導體納米微晶為例,利用CO2-正己烷GXLs,制備由三辛基氧膦(Trioctylphosphine oxide,TOPO)保護的核殼CdSe/ZnS半導體納米量子微晶[51],一次即可完成4個壓力區(qū)間的不同尺寸的量子點分離,分級沉淀出的粒徑分布分別為4.9、3.6、2.3和2.0nm。與反相膠束制備法獲得的CdSe/ZnS微晶平均粒徑4.5nm及傳統(tǒng)合成方法獲得的CdSe/CdS微晶平均粒徑5.2nm相比,GXLs方法的優(yōu)勢在于該過程非常便捷、迅速,操作條件溫和,可在1h內完成,而傳統(tǒng)的液相抗溶劑沉淀法若獲得同樣的目標產(chǎn)物則通常需要幾個小時;另外,GXLs過程不僅所獲得的量子點的粒徑分布非常窄,而且可實現(xiàn)由多分散納米粒徑到不同粒徑分布的單分散顆粒的分級分離。
3.3 存在問題
盡管 GXLs 應用于各種納米材料制備已有不少報道[44,46,47,51],但顯然研究人員更側重于其技術和工藝方面的研究,雖然GXLs可將納米金顆粒的多分散度由26%降至11%,但相應的粒徑標準偏差仍在10%以上;傳統(tǒng)的良溶劑/不良溶劑組合用于納米顆粒的尺寸窄化已可以獲得尺寸分布的相對標準偏差在5%以內,而GXLs目前尚不能做到;此外,對于GXLs 納米材料制備而言,在 GXLs的諸多溶劑性質如組成、密度、粘度、擴散系數(shù)、折射率及介電常數(shù)等中,哪些溶劑參數(shù)對于控制納米微粒的尺寸或形貌修飾最為重要?為什么納米材料的粒徑分布及其重復性與GXLs的壓力區(qū)間及對應過程的時間控制有關?可能的原因有待進一步探討。
3.4 研究趨勢
若想實現(xiàn)GXLs-納米材料可控制備的過程優(yōu)化,有賴于針對目標過程機理的分子水平的研究,有賴于對于過程介質GXLs的穩(wěn)態(tài)及動態(tài)溶劑化的表征。在GXLs溶劑化研究方面,代表性的工作如李宏平和Maroncelli等利用多種先進的實驗手段結合MD模擬,進行了系列 GXLs溶劑化特征的研究[52~55],獲得了GXLs體系的局域組成、粘度、擴散系數(shù)、介電常數(shù)等重要溶劑參數(shù)及介電豫馳時間隨溫度及GXLs表觀組成的定量變化關系;還獲得了GXLs三元體系的優(yōu)先溶劑化、局域環(huán)境及溶劑化動態(tài)學、溶質-溶劑間的相互作用能等信息;發(fā)現(xiàn)GXLs在聚集區(qū)的不均勻性會對其局域傳輸及溶劑性質產(chǎn)生影響,可望利用上述研究結果對GXLs進行設計,使之增強或擬制目標產(chǎn)物的溶解度,預期可為 GXLs 納米制備過程的優(yōu)化設計提供定量依據(jù)。
4 結語
盡管人們對GXLs的了解已有一段時間,但只是在最近幾年科研人員才意識到其潛在而廣泛的應用價值。隨著人們對GXLs性質的了解,在傳統(tǒng)方法基礎上結合GXLs特點的制備技術在材料處理及微粒制備等方面的應用日益得到關注,其工作涉及無機、有機、聚合物、生物醫(yī)學、藥物以及光電納米器件等的制備和性能研究,所采用的技術也拓展到膨脹液體的物理和化學方法的綜合。由于GXLs既有一般有機溶劑的高溶解性能,又有類似于氣體的高擴散、低表面張力及粘度等特點,同時GXLs的許多性質如密度、表面張力、粘度、擴散系數(shù)及溶解性很容易通過壓力及溫度的微小改變而進行調節(jié),所以在材料制備中具備其它技術不具有的優(yōu)點,因此,可以制備一些其它技術難以制備的特殊材料。一些需要降低粘度的工業(yè)化過程已經(jīng)開始了,利用粘度降低特性的未來商業(yè)化應用可能會發(fā)生在聚合物處理及微粒制備方面,可以預期,各種GXLs過程將繼續(xù)在新領域及一些未知領域得到應用。
參考文獻
[1] J P Hallett, C L Kitchens, R Hernandez et al. Acc. Chem. Res., 2006, 39: 531~538.
[2] C A Eckert, C L Liotta, D Bush et al. J. Phys. Chem. B, 2004, 108: 18108~18118.
[3] J C de la Fuente Badilla, C J Peters, J de Swaan Arons. J. Supercrit. Fluids, 2000, 17: 13~23.
[4] C J Chang. J. Chem. Eng. Jpn., 1992, 25: 164~169.
[5] M C Daniel, D Astruc. Chem. Rev., 2004, 104: 293~346.
[6] X F Xie, C L Liotta, C A Eckert. Ind.Eng. Chem. Res., 2004, 43: 2605~2609.
[7] M Wei, G T Musie, D H Busch et al. Green Chem., 2004, 6: 387~392.
[8] G Musie, M Wei, B Subramaniam et al. Coord. Chem. Rev., 2001, 219-221: 789~820.
[9] S G Kazarian, N Sakellarios, C M Gordon. Chem. Commun., 2002, 1314~1315.
[10] H Hammam, B Sivik. J. Supercrit. Fluids, 1993, 6: 223~227.
[11] E Cleve, E Bach, E Schollmeyer. Angew. Makromol. Chem., 1998, 256: 39~48.
[12] Y Kishimoto, R Ishii. Polymer, 2000, 41: 3483~3485.
[13] E Kukova, M Petermann, E Weidner. Chem.-Ing.-Tech., 2004, 76: 280~284.
[14] X Liao, J Wang, G Li et al. J. Polym. Sci., Part B: Polym.Phys., 2004, 42: 280~285.
[15] S L Shenoy, T Fujiwara, K J Wynne. Macromol. Symp., 2003, 201: 171.
[16] Y T Shieh, K H Liu. J. Polym. Sci., Part B: Polym. Phys., 2004, 42: 2479~2489.
[17] Y T Shieh, H S Yang. J. Supercrit. Fluids, 2005, 33: 183~192.
[18] J Jung, M Perrut. J. Supercrit. Fluids, 2001, 20: 179~219.
[19] J Fages, H Lochard, J -J Letourneau et al. Powder Technol., 2004, 141: 219~226.
[20] S D Yeo, E Kiran. J. Supercrit. Fluids, 2005, 34: 287~308.
[21] F Graser,G Wickenhaeuser. BASF: USP 4,451,654, 1982 (CAN 101:92903).
[22] K Kokot, Z Knez, D Bauman. Acta Aliment., 1999, 28: 197~208.
[23] S M Pourmortazavi, S S Hajimirsadeghi. Ind. <st1:country-region w:st="on">Eng. Chem. Res., 2005, 44: 6523~6533.
[24] A Bertucco. In: P G Jessop, W Leitner, Eds. Chemical Synthesis Using Supercritical Fluids. Weinheim, <st1:country-region w:st="on">Germany: Wiley-VCH; 1999.
[25] S Palakodaty, P York. Pharm. Res., 1999, 16: 976~976.
[26] P G Jessop, B Subramaniam. Chem. Rev., 2007, 107: 2666~2694.
[27] P Sencar-Bozic, S Srcic, Z Knez et al. Int. J. Pharm., 1997, 148: 123~130.
[28] A Shine, J Gelb. WO Patent 98/15348, 1998.
[29] V J Krukonis, P M Gallagher, M P Coffey. USP 5,360,478, 1994 (application 1991, CAN 122:34594).
[30] C N Field, P A Hamley, J M Webster et al. J. Am. Chem. Soc., 2000, 122: 2480~2488.
[31] A O’Neil, C Wilson, J M Webster et al. Angew. Chem., Int. Ed. 2002, 41: 3796~3799.
[32] J Bleich, B W Müller, W Wassmus. Int. J. Pharm., 1993, 97: 111~117.
[33] G J Hutchings, J K Bartley, J M Webster et al. J. Catal., 2001, 197: 232~235.
[34] C A Johnson, S Sharma, B Subramaniam et al. J. Am. Chem. Soc., 2005, 127: 9698~9699.
[35] M Hanna, P York. WO Patent 95/01221 A1, 1994 (AN 2005: 769844).
[36] B Subramaniam, S Saim, R A Rajewski et al. In: P T Anastas, L G Heine, T C Williamson, Eds. Green Engineering. Washington, D C: Oxford University Press; 2000.
[37] W K Snavely, B Subramaniam, R A Rajewski et al. J. Pharm. Sci., 2002, 91: 2026~2039.
[38] F Niu, K F Roby, R A Rajewski et al. In: S Svenson, Ed. Polymeric Drug Delivery Volume II - Polymeric Matrices and Drug Particle Engineering. Washington, DC: Oxford University Press; 2006.
[39] M Gimeno, N Ventosa, S Sala et al. Cryst. Growth Des., 2006, 6: 23~25.
[40] J Chen, J L Zhang, D X Liu et al. Colloids Surf., B, 2004, 33: 33~37.
[41] H F Zhang, J Lu, B X Han. J. Supercrit. Fluids, 2001, 20: 65~71.
[42] J L Zhang, B X Han, J C Liu et al. Chem. Commun., 2001, 2724~2725.
[43] J Zhang, B Han, J Liu et al. Chemistry-A Eur. J., 2002, 8: 3879~3882.
[44] M C McLeod, C L Kitchens, C B Roberts. Langmuir, 2005, 21: 2414~2418.
[45] J Liu, M Anand, C B Roberts. Langmuir, 2006, 22: 3964~3971.
[46] M Anand, M C McLeod, P W Bell. J. Phys. Chem. B, 2005, 109: 22852~22859.
[47] M C McLeod, M Anand, C L Kitchens. Nano Lett., 2005, 5: 461~465.
[48] B Subramaniam, S Saim, R Rajewski et al. USP 5,833,891, 1998 (CAN 129:347344).
[49] D Liu, J Zhang, B Han et al. Colloids Surf., A 2003, 227: 45~48.
[50] J Zhang, B Han, J Liu et al. J. Supercrit. Fluids, 2004, 30: 89~95.
[51] M Anand, L A Odom, C B Roberts. Langmuir, 2007, 23: 7338~7343.
[52] H Li, M Maroncelli. J. Phys. Chem. B, 2006, 110: 21189~21197.
[53] H Li, S Arzhantsev, M Maroncelli. J. Phys. Chem. B, 2007, 111: 3208~3221.
[54] C Swalina, S Arzhantsev, H Li et al. J. Phys. Chem. B, 2008, 112: 14959~14970.
[55] H Li, L Ding, Q Xu et al. World Sci-Tech R &D 2007, 29: 19~31.
注:本文為提供者翻譯的,由于知識所限,其中錯誤在所難免,敬請原諒。如有問題可以查找原文。
統(tǒng)計數(shù)據(jù)
共收錄化學品數(shù)據(jù)
147579 條
公告
更多>
-
微信掃一掃
關注:物競化學品數(shù)據(jù)庫
-
物竟數(shù)據(jù)庫 手機版
國內首款化工類專業(yè)手機應用
-
微博賬號
wjhxp
快速導航
化學品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
關于物競
物競數(shù)據(jù)庫是一個全面、專業(yè)、專注,并且免費的中文化學品信息庫,為學生、學者、化學品研究機構、檢測機構、化學品工作者提供專業(yè)的化學品平臺進行交流。
數(shù)據(jù)庫采用全中文化服務,完全突破了中英文在化學物質命名、化學品俗名、學名等方面的差異,所提供的數(shù)據(jù)全部中文化,更方便國內從事化學、化工、材料、生物、環(huán)境等化學相關行業(yè)的工作人員查詢使用。
關注我們
-
微信賬號:物競化學品數(shù)據(jù)庫

-
微博賬號:wjhxp
聯(lián)系我們
上海市延長路149號上海大學科技園412室
公司總機: 021-56389801
訂購電話: 4007001514
傳真電話: 021-56389802
客服電話: 021-56332350
電子郵件: wingch@basechem.org
Copyright ? 物競數(shù)據(jù)庫 2009-2026.All Rights Reserved. 關于數(shù)據(jù)庫 | 關于物競 | 法律聲明 | 熱門關鍵字
 滬公網(wǎng)安備 31010602001115號
滬公網(wǎng)安備 31010602001115號