脂質(zhì)體毛細(xì)管電泳研究進(jìn)展
- 2012-05-15
- 專題
自1965年由英國(guó)的Bangham等(1)首先發(fā)現(xiàn)磷脂在水中可以自發(fā)形成脂質(zhì)體(Liposome)以來(lái),對(duì)脂質(zhì)體的實(shí)驗(yàn)研究日漸廣泛,已遍及生命科學(xué)、膜工程學(xué)等領(lǐng)域,并逐漸向臨床應(yīng)用發(fā)展。脂質(zhì)體是由磷脂和其它兩親性物質(zhì)分散于水中形成的由一層或多層同心脂質(zhì)雙分子膜包封而成的球狀體,具有親水性和疏水性兩性性質(zhì)。依其所含脂質(zhì)雙分子層的層數(shù),脂質(zhì)體可分為單室脂質(zhì)體和多室脂質(zhì)體。其中單室脂質(zhì)體又可根據(jù)粒徑的大小分為小單室脂質(zhì)體(粒徑范圍0.02 ~0.08 μm)和大單室脂質(zhì)體(粒徑范圍0.1~1μm)。
隨著生物膜色譜(Biomembrane Chromatography)的發(fā)展,脂質(zhì)體作為一種分析工具受到了越來(lái)越多的關(guān)注,并成為近年來(lái)較為活躍的研究領(lǐng)域(2,3)。生物膜色譜是一種新型液相色譜模式,它以天然或人工模擬生物膜為固定相,主要用來(lái)研究溶質(zhì)分子與生物膜間的相互作用。目前,最常用的生物膜色譜固定相是固定化脂質(zhì)體(4-6),因?yàn)樗芨玫啬M生物膜的脂雙層結(jié)構(gòu),并具備生物膜的流動(dòng)性特征。這種人工模擬的生物膜體系不僅可以精確地模擬生物膜的化學(xué)環(huán)境,而目其物理性能也可以通過(guò)調(diào)節(jié)溫度、pH、離子強(qiáng)度等得到有效控制。
固定化脂質(zhì)體色譜具有許多優(yōu)點(diǎn),但是它的應(yīng)用并不廣泛,主要原因是制柱比較困難、成本高。其次高效液相色譜(High Performance Liquid Chromatography ,HPLC)常用的硅膠基質(zhì)及其化學(xué)鍵合固定相的pH適用范圍較窄(2~8),色譜柱易被污染且難再生,流動(dòng)相需要大量有機(jī)溶劑,成本較高,且污染環(huán)境。與固定化脂質(zhì)體色譜相比,脂質(zhì)體毛細(xì)管電泳(Liposome Capillary Electrophoresis ,LCE)更有優(yōu)勢(shì),其具有制備簡(jiǎn)便、溶劑消耗少、進(jìn)樣量小、成本低等優(yōu)點(diǎn)。1995年Zhang等(7)首次提出了LCE的概念,近年來(lái),LCE已經(jīng)得到了較快發(fā)展,尤論是在方法學(xué)方面,還是在應(yīng)用方面都有了比較深入的研究。
1脂質(zhì)體的制備
目前,脂質(zhì)體的制備技術(shù)己經(jīng)相當(dāng)成熟,有關(guān)報(bào)道和綜述也較多(8-10)。常用脂質(zhì)體制備方法有薄膜分散法、注入法、超聲分散法、冷凍干燥法、凍融法、逆相蒸發(fā)法、復(fù)乳法等。
LCE中所用的脂質(zhì)體主要是單室脂質(zhì)體。多室脂質(zhì)體由于背景噪聲大,靈敏度低而不適合。與固定化脂質(zhì)體色譜相比,LCE中的脂質(zhì)體制備方法比較單一,通常采用薄膜-超聲分散法和薄膜-擠壓分散法。薄膜-超聲分散法是制備單室脂質(zhì)體的最早方法,即將磷脂、膽固醇等類脂質(zhì)溶于有機(jī)溶劑中,旋轉(zhuǎn)蒸發(fā)除去有機(jī)溶劑,使在燒瓶?jī)?nèi)壁形成薄膜,然后將水溶性介質(zhì)加入燒瓶中并不斷振蕩,得到多室脂質(zhì)體,最后經(jīng)超聲處理得到單室脂質(zhì)體,其粒徑大小依超聲時(shí)間的長(zhǎng)短而不同。薄膜-超聲分散法的缺點(diǎn)是其制備的脂質(zhì)體粒徑分布不太均勻,內(nèi)層的磷脂分子數(shù)要小于外層的磷脂分子數(shù),熱力學(xué)不穩(wěn)定,脂質(zhì)體之間容易發(fā)生融合(11)。薄膜-擠壓分散法是目前制備單室脂質(zhì)體的最常用方法。這種方法也是先采用薄膜法制得多室脂質(zhì)體,然后在一定壓力(<7.091×l05Pa)下使大多室脂質(zhì)體通過(guò)很小孔徑的聚碳酸醋膜,以獲得大小均勻的單室脂質(zhì)體(12)。
2 LCE的分類及脂質(zhì)體涂層的制備
LCE大體可分為兩類。一類從電泳緩沖液入手,將脂質(zhì)體作為添加劑,直接加入電泳緩沖液中,形成假固定相,其基本原理與膠束電動(dòng)毛細(xì)管色譜(Micellar Electroliinetic Capillary Chromatography,MECC)類似,因而被稱之為“脂質(zhì)體電動(dòng)毛細(xì)管色譜(Liposome Electrokinetic CapillaryChromatography LECC)" (7,13~21)。另一類從毛細(xì)管管壁入手,將脂質(zhì)體作為涂層劑,采用氫鍵吸附法(22~27)、靜電吸附法(28,29)、抗生物素蛋白-生物素親和法(30)等固定化方法,使毛細(xì)竹內(nèi)壁形成較穩(wěn)定的脂質(zhì)體涂層,因而可稱之為“固定化脂質(zhì)體毛細(xì)竹電泳(Immobilized Liposome Capillary Electrophoresis, ILCE) "。兩者相比,ILCE的柱效較低,但其脂質(zhì)體的消耗少,并可與質(zhì)譜聯(lián)用(2)。
ILCE的脂質(zhì)體涂層是磷脂雙層(涂層在毛細(xì)管內(nèi)壁的脂質(zhì)體破裂,相互融合)還是囊泡層(涂層在毛細(xì)管內(nèi)壁的脂質(zhì)體形態(tài)完整),取決于多種因素如脂質(zhì)體的制備方法、粒徑大小、載體材料及濃度、電泳緩沖液中Ca2+存在與否等(11,31,32)。脂質(zhì)體涂層的穩(wěn)定性與固定化方法有很大關(guān)系。氫鍵吸附法、靜電吸附法等非共價(jià)鍵合的方法,操作過(guò)程簡(jiǎn)單,但脂質(zhì)體涂層穩(wěn)定性差,流失嚴(yán)重;親和法則使脂質(zhì)體涂層的穩(wěn)定性得到明顯改善,缺點(diǎn)是制作成本相對(duì)較高。下面對(duì)這些固定化方法進(jìn)行一一介紹。
2.1氫鍵吸附法
該法十分簡(jiǎn)便,只需將含有脂質(zhì)體的電泳緩沖液與毛細(xì)管平衡數(shù)分鐘,此時(shí)由于脂質(zhì)體中磷脂的一P=O基團(tuán)與融熔石英毛細(xì)管內(nèi)壁的硅烴基產(chǎn)生氫鍵作用力,而使脂質(zhì)體充分吸附于毛細(xì)竹內(nèi)壁,然后用不含脂質(zhì)體的電泳緩沖液沖洗,除去未吸附的脂質(zhì)體即可。但是對(duì)于負(fù)性脂質(zhì)體而言,該法不太穩(wěn)定,因?yàn)槊?xì)管內(nèi)壁的硅烴基會(huì)與負(fù)性脂質(zhì)體發(fā)生相互排斥作用。
Hautala等(22)選用平均粒徑大約100nm的大單室負(fù)性脂質(zhì)體(組成成分:1-棕相酞-2-油酸-3-磷脂酞膽堿/磷脂酞妊氨酸/膽固醇,POPC/PS/Cholesterol),通過(guò)氫鍵吸附法制備了LCE的脂質(zhì)體涂層。具體涂層過(guò)程為:0.5mol/L鹽酸沖洗裸露熔融石英毛細(xì)管10min→水沖洗15min→電泳緩沖液沖洗5min→含有脂質(zhì)體的電泳緩沖液沖洗10min→含有脂質(zhì)體的電泳緩沖液在毛細(xì)竹內(nèi)預(yù)穩(wěn)定15min→電泳緩沖液沖洗除去未吸附的脂質(zhì)體。在整個(gè)涂層過(guò)程中,電泳緩沖液的種類保持相同。Hautala等發(fā)現(xiàn),脂質(zhì)體涂層的最終效果和穩(wěn)定性受到?jīng)_洗脂質(zhì)體溶液的時(shí)間、分析電壓、電泳緩沖液的種類等因素的影響,其中電泳緩沖液的種類對(duì)負(fù)性脂質(zhì)體涂層的涂層效果影響最大;采用三(三烴甲基氨基甲烷)緩沖液、磷酸欲緩沖液和麥黃酮(N-三烴甲基甲基甘氨酸)緩沖液制備的脂質(zhì)體涂層,電滲流((EOF)有少許增加(1%~7%),而用HEPES [N-(2-羥乙基)哌嗪-N-(2-乙磺酸)]緩沖液制備的脂質(zhì)體涂層,EOF減少24%,這表明在HEPES緩沖液中,脂質(zhì)體與毛細(xì)竹內(nèi)壁的作用最強(qiáng),因而得到最好的涂層效果。Wiedmer等(27)嘗試用一系列與HEPES結(jié)構(gòu)相似的化合物(均含呱嗦基團(tuán))溶液作為電泳緩沖液,結(jié)果發(fā)現(xiàn)采用這些緩沖液制備的負(fù)性脂質(zhì)體(組成成分:磷脂酞膽堿((PC)/PS)涂層也相當(dāng)穩(wěn)定。另外,在電泳緩沖液中加入一價(jià)金屬離了(如Ca2+,Mg2+,Zn2+)或電泳溫度在相變溫度以下時(shí)均能增加磷脂膜的剛性,提高脂質(zhì)體涂層的穩(wěn)定性(24,25)。
2.2靜電吸附法
先將帶電荷的化合物涂布于毛細(xì)竹內(nèi)壁,然后將帶相反電荷的脂質(zhì)體填充于毛細(xì)竹中,則脂質(zhì)體依靠靜電作用力吸附于毛細(xì)竹內(nèi)壁。因?yàn)殪o電作用力比氫鍵作用力大,所以這種方法制備的脂質(zhì)體涂層比氫鍵吸附法制備的脂質(zhì)體涂層更穩(wěn)定一些。
Ornskov等(30)先將瓊脂糖衍生物(含有帶正電荷的銨離了)涂布于毛細(xì)竹內(nèi)壁,然后用含有負(fù)性脂質(zhì)體(組成成分:POPC/PS)的溶液沖洗毛細(xì)管5min,則負(fù)性脂質(zhì)體通過(guò)靜電作用力吸附在帶正電荷的毛細(xì)管內(nèi)壁。實(shí)驗(yàn)發(fā)現(xiàn),隨著涂布于毛細(xì)管內(nèi)壁的瓊脂糖衍生物電荷密度的增加,固定在毛細(xì)竹內(nèi)壁的脂質(zhì)體含量增大,一系列非離子化合物在ILCE中的保留值增大。這證明在脂質(zhì)體的涂層過(guò)程中,靜電作用力確實(shí)起主要作用。
2. 3抗生物素蛋白一生物素親和法
首先將抗生物素蛋白共價(jià)鍵合到氨基化的毛細(xì)竹內(nèi)壁,然后將生物素酞基化的脂質(zhì)體填充到鍵合有抗生物素蛋白的毛細(xì)管內(nèi)(如圖1)。通過(guò)抗生物素蛋白-生物素的親和作用力將脂質(zhì)體固定在毛細(xì)管內(nèi)表面。由于溶解脂質(zhì)體的電泳緩沖液中含有抗生物素蛋白,因而單個(gè)生物素酞基化的脂質(zhì)體之間也可通過(guò)親和作用力發(fā)生聚合。與氫鍵吸附法制備的脂質(zhì)體涂層相比,親和法制備的脂質(zhì)體涂層其穩(wěn)定性有了相當(dāng)程度的提高,因?yàn)榭股锼氐鞍着c生物素間的親和作用力比氫鍵作用力強(qiáng)得多。在涂層過(guò)程中還發(fā)現(xiàn),毛細(xì)管內(nèi)壁覆蓋了4~15層脂質(zhì)體,且采用大單室脂質(zhì)體制備的脂質(zhì)體涂層所固定的磷脂含量要比采用小單室脂質(zhì)體制備的脂質(zhì)體涂層所固定的磷脂含量高出2倍。

3 LCE的應(yīng)用
LCE的應(yīng)用的應(yīng)用領(lǐng)域主要分為二個(gè)方面:(1)在各種有機(jī)化合物及多膚、蛋白質(zhì)分離中的應(yīng)用;(2)在對(duì)映異構(gòu)體分離中的應(yīng)用;(3)在藥物一生物膜相4.作用研究中的應(yīng)用。
3.1 LCE在各種有機(jī)化合物及多膚、蛋白質(zhì)分離中的應(yīng)用
LCE中,溶質(zhì)的分離主要基于不同溶質(zhì)在電泳緩沖液和脂質(zhì)體 (LECC中為假固定相,ILCE中為涂層)之間的分配系數(shù)不同而進(jìn)行的。溶質(zhì)的帶電量、化學(xué)結(jié)構(gòu)、極性、甚至濃度都會(huì)影響溶質(zhì)的分配系數(shù)大小(33,34)。另外,在分配過(guò)程中,不同溶質(zhì)在脂質(zhì)體中的分配部位也可能不同(極性區(qū)域或疏水區(qū)域) (35)。
目前,LCE已用于苯的衍生物及苯酚類化合物(14,15,18,25)、類固醇(15~17,22,25)、多膚和蛋白質(zhì)(7,19~21,23,24,28)、β-阻斷劑(18)、其它藥物(7,13,18,26,27)等的分離。Wiedmer等(16,17)在電泳緩沖液中添加負(fù)性脂質(zhì)體分離了8種中性皮質(zhì)類固醇化合物(17-異醛固酮、醛固酮、21-脫氧皮質(zhì)醇、皮質(zhì)酮、雄烯二酮、睪酮、黃體酮和17-烴孕酮)。脂質(zhì)體中總脂質(zhì)的含量、脂質(zhì)體組成中不同磷脂的摩爾比、脂質(zhì)頭部基團(tuán)的種類、電泳緩沖液的種類以及脂質(zhì)體的物理狀態(tài)(膠態(tài)和液品態(tài))等多種因素都會(huì)影響分離效果(16)。Wiedmer等(17)分離衡體激素時(shí)還發(fā)現(xiàn),脂質(zhì)體組成成分中膽固醇的含量對(duì)類固醇化合物的分離有很大影響;膽固醇含量越高,化合物的分離度越大。
CE分離分析多膚和蛋白質(zhì)時(shí),樣品的吸附嚴(yán)重影響了分離分析結(jié)果的重現(xiàn)性。這種吸附主要是帶部分正電荷的多膚和蛋白質(zhì)分子與帶負(fù)電荷的熔融石英毛細(xì)竹內(nèi)壁相互作用的結(jié)果。而LCE分離分析多膚和蛋白質(zhì)時(shí),由于毛細(xì)竹內(nèi)壁吸附了脂質(zhì)體,硅烴基被掩蔽,樣品的吸附減少,因而使柱效增大,電泳峰形和分離分析結(jié)果的重現(xiàn)性明顯改善;同時(shí),多肽、蛋白質(zhì)與脂質(zhì)體之間的相互作用使分離選擇性得到提高(19,20,23)。 Corradini等(19,20)在電泳緩沖液中添加POPC脂質(zhì)體分別分離了4種堿性蛋白(細(xì)胞色素C,溶菌酶,核糖核酸酶A, a-胰凝乳蛋白酶原)和3種合成多膚(血管緊縮素I,Ⅱ,Ⅲ);研究發(fā)現(xiàn),毛細(xì)竹內(nèi)注入的脂質(zhì)體溶液的體積或脂質(zhì)體載體材料(磷脂)的濃度增加,多膚的遷移時(shí)間都增加,分離度提高(20)。Cunliffe等(23)采用ILCE(脂質(zhì)體組成:二月桂酞磷脂酞膽堿/Ca2+,DLPC/Ca2+),在pH3~10范圍內(nèi)成功分離了陰性和陽(yáng)性蛋白質(zhì),理論塔板數(shù)高達(dá)1400000/m遷移時(shí)間的日內(nèi)精密度小于1.3%,日間精密度小于4%,回收率達(dá)到93%以上。
3.2 LCE在對(duì)映異構(gòu)體分離中的應(yīng)用
在對(duì)映異構(gòu)體的CE分離中,一般采用在電泳緩沖液中加入乎性試劑的方法。很多天然生物大分子如牛血清蛋白、人血清蛋白、糖蛋白、溶菌酶等本身含有不對(duì)稱碳原子,能夠?qū)υS多小分子化合物產(chǎn)生乎性識(shí)別作用,是一類高效、廉價(jià)、易得的乎性試劑(36)。但是這類天然生物大分子在紫外光區(qū)都有強(qiáng)烈的吸收,因而大大限制了其應(yīng)用范圍。將這類乎性試劑固定在毛細(xì)竹內(nèi)壁是解決此問(wèn)題的主要方法。Bo等(28)首次將溶菌酶固定在毛細(xì)竹內(nèi)壁脂質(zhì)體涂層的磷脂膜上,在電泳緩沖液中不含乎性試劑的情況下,成功分離了D-和L-色氨酸。他們?cè)谕繉舆^(guò)程中發(fā)現(xiàn),若將負(fù)性脂質(zhì)體直接涂敷在裸露的毛細(xì)竹內(nèi)壁,則涂層的穩(wěn)定性很差。為解決該問(wèn)題,Bo等先在毛細(xì)竹內(nèi)壁涂敷M1C4(1-(4-碘丁基)-1,4-甲基哌嗪-1-碘化銨),然后再涂敷負(fù)性脂質(zhì)體,最后用溶菌酶溶液沖洗涂層后的毛細(xì)竹。因?yàn)?M1C4掩蔽了帶負(fù)電荷的硅經(jīng)基,減少了負(fù)性脂質(zhì)體與硅經(jīng)基的排斥作用,而目與負(fù)性脂質(zhì)體靜電相吸,所以脂質(zhì)體涂層的穩(wěn)定性大大提高。溶菌酶由于較易滲入到磷脂膜內(nèi),而被比較穩(wěn)定地固定于毛細(xì)竹內(nèi)壁。與此同時(shí),Bo等還嘗試將M1C4涂敷于毛細(xì)竹內(nèi)壁后直接涂敷溶菌酶,雖然D-和L-色氨酸的分離度提高了,但卻以犧牲穩(wěn)定性為代價(jià),分離結(jié)果的重現(xiàn)性很差。
3.3 LCE在藥物與生物膜相互作用研究中的應(yīng)用
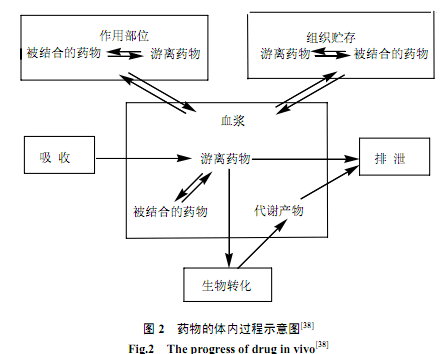
藥物在體內(nèi)的吸收、分布和排泄統(tǒng)稱為藥物轉(zhuǎn)運(yùn),而在體內(nèi)發(fā)生的化學(xué)變化,則稱為藥物代謝或生物轉(zhuǎn)化。在這些過(guò)程中,各種因素的相互影響如圖2所示(37)。因?yàn)槿梭w內(nèi)的各部分之間彼此有生物膜隔開(kāi),所以生物膜是影響藥物體內(nèi)過(guò)程的最主要因素之一。藥物的活性、毒性、體內(nèi)分布及其它生理過(guò)程都與藥物在生物膜上的分配狀況相關(guān)(38),因此測(cè)定藥物在生物膜上的分配系數(shù)對(duì)藥物設(shè)計(jì)和藥物篩選來(lái)說(shuō)十分重要。由于LCE提供了一個(gè)人工模擬的生物膜表面,能夠模擬藥物結(jié)合到生物膜表面或分配到生物膜脂雙層時(shí)發(fā)生的疏水、靜電等相互作用,因而藥物在LCE中的遷移行為和選擇性能夠直接反映藥物在生物膜上的分配狀況。
藥物在LCE中的遷移行為取決于EOF、藥物與脂雙層的相互作用、磷脂(或其它脂類尤其是膽固醇與磷脂的比例)的用量等諸多因素。為了表示藥物與脂雙層的相互作用,可參照色譜原理,假定一個(gè)與EOF和磷脂的用量無(wú)關(guān)的容量因子(見(jiàn)式((1))(7)。
k'=(t- t') /( t'×B) (1)
式中k'為容量因子;t為藥物在脂質(zhì)體毛細(xì)竹電泳中的遷移時(shí)間;t'為藥物在無(wú)脂質(zhì)體毛細(xì)竹電泳中的遷移時(shí)間;B為磷脂的用量。
通過(guò)式(2)還可以將藥物在LCE中的容量因子k'與藥物的脂質(zhì)體一水分配系數(shù)K1w聯(lián)系起來(lái)。
k'=φK1w (2)
式中φ為相比,等于LCE中脂質(zhì)體與電泳緩沖液所占的體積比。
一個(gè)藥物在生物膜上的分配情況通常用該藥物在正辛醇和水中濃度比值的對(duì)數(shù)(lgPow)來(lái)衡量(19,39)。然而正辛醇與生物膜有很大差異,用lgPow評(píng)價(jià)藥物與生物膜的相互作用是不夠準(zhǔn)確的。脂質(zhì)體的結(jié)構(gòu)與生物膜很相似,因此,K1w能更準(zhǔn)確地反映藥物與生物膜的相互作用。
由式(2)可知,K1w與相比φ有關(guān)。在LCE中,φ是一個(gè)反映脂質(zhì)體本質(zhì)特征的參數(shù),當(dāng)脂質(zhì)體的組成恒定時(shí),φ不變,且能較準(zhǔn)確地計(jì)算出來(lái)。在HPLC中,φ是固定相與流動(dòng)相所占的體積比,因而不同的色譜柱,φ的差異性很大,且不易計(jì)算(18)。因此,與固定化脂質(zhì)體色譜相比,采用LCE測(cè)定K1w更準(zhǔn)確,重現(xiàn)性更好。
Burns等(14,18)首次將LCE技術(shù)應(yīng)用于定量測(cè)定藥物的K1w。研究表明,LCE測(cè)定的K1w與lgPow、線性溶劑化能量關(guān)系式(Linear Solvation Energy Relationship, LSER}是定量結(jié)構(gòu)分配關(guān)系的一種表現(xiàn)形式)所預(yù)測(cè)的化合物的K1w值都有很好的相關(guān)性。這說(shuō)明脂質(zhì)體的溶劑化特性與正辛醇相似,并且K1w還可應(yīng)用于定量結(jié)構(gòu)分配關(guān)系(Quantitative Structure-Partition Relationship, QSPR)方面的研究。
藥物的吉布斯自由能變化△Go即藥物在無(wú)脂質(zhì)體毛細(xì)竹內(nèi)的吉布斯自由能與在脂質(zhì)體毛細(xì)竹內(nèi)的吉布斯自由能之差,是評(píng)價(jià)藥物與生物膜間相互作用的另一個(gè)重要參數(shù)(7,13,26)。△Go越小,說(shuō)明藥物與生物膜間的相互作用越強(qiáng),藥物更易進(jìn)入生物膜內(nèi)。但△Go的計(jì)算公式比較復(fù)雜,為此,通常選擇一種參比藥物(如阿司匹林),并假設(shè)EOF為零,通過(guò)計(jì)算0(OCe>見(jiàn)式(3)來(lái)衡量藥物的△Go(7)。
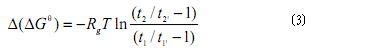
式中Rg為標(biāo)準(zhǔn)氣體常數(shù);T為熱力學(xué)溫度;t1為參比藥物在脂質(zhì)體毛細(xì)管電泳中的遷移時(shí)間;t1’為參比藥物在無(wú)脂質(zhì)體毛細(xì)管電泳中的遷移時(shí)間;t2為被測(cè)藥物在脂質(zhì)體毛細(xì)竹電泳中的遷移時(shí)間;t2’為被測(cè)藥物在無(wú)脂質(zhì)體毛細(xì)竹電泳中的遷移時(shí)間。
Manetto等(13,26)分別采用LECC和ILCE(脂質(zhì)體組成成分:POPC)測(cè)定了一系列不同極性藥物的△(△Go),對(duì)這些藥物與脂質(zhì)體間的相互作用進(jìn)行了探討,并比較了兩種方法測(cè)定結(jié)果的差異性。在LECC中,他們選用聚酞亞胺涂層毛細(xì)竹(總長(zhǎng)30cm,有效長(zhǎng)度19.5cm,內(nèi)徑50μm),樣品從負(fù)極端進(jìn)樣;電泳緩沖液pH9.2時(shí),水楊酸、阿司匹林、酮基布洛芬、苯妥英和普蔡洛爾5種藥物全部與脂質(zhì)體發(fā)生相互作用,但測(cè)得的水楊酸的△(△Go)值比文獻(xiàn)[7](采用LECC)測(cè)得的水楊酸的△(△Go)值低0.2 kJ/mol(約17%) ,可能因?yàn)殡娪揪彌_液pH9.2時(shí)仍有殘留的EOF而導(dǎo)致測(cè)定結(jié)果偏低,因?yàn)榭鄢鼸OF后計(jì)算得到的△(△Go)與文獻(xiàn)[7]的測(cè)得值很接近(13)。在ILCE中,選用未涂層的毛細(xì)竹,測(cè)得的水楊酸的從△(△Go)值(扣除了EOF的影響)比LECC測(cè)得的值高出5%(26)。
一般來(lái)說(shuō),在利用LCE研究藥物與生物膜的相互作用時(shí),為了使脂質(zhì)體與人體生物膜具有更大的相似性,電泳緩沖液pH的選擇應(yīng)依據(jù)相應(yīng)的人體生理pH(如小腸中生理pH約5.4,細(xì)胞外生理pH約7.0,細(xì)胞內(nèi)生理pH7.4)而定。另外,由于pH會(huì)影響脂質(zhì)體載體材料的水解速度(pH過(guò)大或過(guò)小,磷脂的水解速度都將增大(40),因此LCE應(yīng)使用新鮮制備的脂質(zhì)體。
4展望
LCE作為一種新穎的分離分析方法,目前僅有相關(guān)的研究報(bào)道。LCE除在分離分析方面具有一定潛力外,更重要的是脂質(zhì)體與生物膜結(jié)構(gòu)類似,利用LCE研究藥物與脂質(zhì)體間的相互作用,能最大程度地反映藥物分子與生物膜相互作用的真實(shí)情況。這種體外實(shí)驗(yàn)?zāi)M體內(nèi)條件的篩選模型為藥物篩選和藥物設(shè)計(jì)提供了強(qiáng)有力的工具。再者,LCE與生物膜色譜相比,方法簡(jiǎn)單,可操作性強(qiáng),成本低廉,尤其是測(cè)定藥物的K1w準(zhǔn)確、重現(xiàn)性好,具有更好的發(fā)展前景。目前,LCE無(wú)論在理論還是技術(shù)方面都還不太成熟,應(yīng)用范圍也較為有限,因而其發(fā)展空間還很廣闊。
參考文獻(xiàn)
(1)A D Bangham, MM Standisli, J C Watkins. J. Mol. Biol., 1965, 13:238252
(2)A C6mez-Hens, J M Femandez-Romero. Trends in Ana1. Chem,2005,24(1):919
(3)S K Wiedmer, M-L Riekkola, M S Jussila. Trends in Anal. Chem., 2004, 23(8):562582
(4)Q Yang, X-Y Liu, M Yoshimoto et al. Anal. Biocliem,1999, 268:354362
(5)Q Yang, X-Y Liu, S-I Ajiki et al. J. Chromatogr. B, 1998, 707:131141
(6)E Kranse, M Dathe, T Wieprecht et al. J. Chromatogr. A, 1999, 849:125133
(7)Y X Zhang, R Zhang, S Hjeren et al. Electrophoresis, 1995, 16:15191523
(8)胡景蓮,張輝.中國(guó)臨床醫(yī)藥研究雜志.2004, 113:1187211874
(9)曹寧寧,劉金鵬.天津理工學(xué)院學(xué)報(bào),2003, 19(1):3035
(10)趙海霞,郭興奎,孔德亮等.山東中醫(yī)雜志,2000, 19(7):435437
(11)I Reviakine, A Brisson. Langmuir, 2000, 16:1806}}1815
(12)高申.現(xiàn)代藥物新劑型新技術(shù).北京:人民軍醫(yī)出版社,2002:213
(13)G Manetto, M S Bellini, Z J Deyl. J. Cbromatogr. A, 2003, 990:205214
(14)S T Bums, A A Agbodjan, M G Khaledi. J. Cbromatogr. A, 2002, 973:167176
(15)S K Wiedmer, J Hautala, J M Holopainen et al. Electrophoresis, 2001, 22:13051313
(16)S K Wiedmer, J M Holopainen, P Mustakangas et al. Electrophoresis, 2000, 21:31913198
(17)S K Wiedmer, M S Jussila, J M Holopainen et al. J. Sep. Sci, 2002, 25(7):427437
(18)S T Bums, M G Kbalehi. J. Pbann. Sci, 2002, 91(7):16011612
(19)D Corradini, G Mancini, C Bello. J. Chromatogr. A, 2004, 1051:103110
(20)D Corradini, G Mancini, C Bello. Chromatogr. Supplement, 2004, 60:S 125}S 132
(21)C Bello, G Mancini, D Corradini. Cbimica a 1'Industria (Milan, <st1:country-region w:st="on">Italy), 2004, 86(7):7883
(22)J T Hautala, M V Linden, S K Wiedmer et al. J. Chromatogr. A, 2003, 1004:8190
(23)J M Cunliffe, N E Baryla, C A Lucy. Aual. Chem,2002, 74:776783
(24)M V Linden, S K Wiedmer, R M S Hakala et al. J. Chromatogr. A, 2004, 1051:6168
(25)J T Hautala, S K Wiedmer, M-L Riekkola. Anal. Bioanal. Chem., 2004, 378(7):17691776
(26)G Manetto, M S Bellini, Z Deyl. J Chromatogr. A, 2003, 990:281289
(27)S K Wiedmer, M Jussila, R M S Hakala et al. Electrophoresis, 2005, 26:19201927
(28)T Bo, S K Wiedmer, M-L Riekkola. Electrophoresis, 2004, 25:17841791
(29)E ornskov, S Ullsten, L Soderberg et al. Electrophoresis, 2002, 23(19):33813384
(30)Q Yang, X-YLin, JMiyake. Supramol. Sci,1998, 5(5-6): 769772
(31)R Rapuano, A M Carmona-Ribeiro. J. Colloid Interface Sci., 1997, 193: 104111
(32)A S Muresan, K Y C Lee. J. Pbys. Chem. B, 2001, 105: 852855
(33)M Alumed, J S Burton, J Hadgraft et al. Biochem. Pbarmacol., 1980, 29: 23612365
(34)K Kitamura, H Kano, K Yoneyama. Mol. Phannacol., 1981, 20:124127
(35)A M S Ahmed, F H Farab, I W Kellaway. Pharm. Res., 1985, 2:119124
(36)J Haginaka. J. Cbromatogr. A, 2001, 906:253273
(37)王欽茂.藥理學(xué).上海:上海科技出版社,1994:265
(38)C Pidgeon, S Ong, H-L Liu et al. J. Med. Chem., 1995, 38: 590594
(39)郭宗儒.藥物化學(xué)總論.北京:中國(guó)醫(yī)藥利一技出版社,2003:171
(40)M Grit, W J M Underberg, D J A Crommelin. J. Pbarm. Sci., 1993, 82(4): 362366
注:本文為提供者整理翻譯,由于知識(shí)所限,其中錯(cuò)誤在所難免,敬請(qǐng)?jiān)彙H缬袉?wèn)題可以查找原文。
統(tǒng)計(jì)數(shù)據(jù)
共收錄化學(xué)品數(shù)據(jù)
147579 條
公告
更多>
-
微信掃一掃
關(guān)注:物競(jìng)化學(xué)品數(shù)據(jù)庫(kù)
-
物竟數(shù)據(jù)庫(kù) 手機(jī)版
國(guó)內(nèi)首款化工類專業(yè)手機(jī)應(yīng)用
-
微博賬號(hào)
wjhxp
快速導(dǎo)航
化學(xué)品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
關(guān)于物競(jìng)
物競(jìng)數(shù)據(jù)庫(kù)是一個(gè)全面、專業(yè)、專注,并且免費(fèi)的中文化學(xué)品信息庫(kù),為學(xué)生、學(xué)者、化學(xué)品研究機(jī)構(gòu)、檢測(cè)機(jī)構(gòu)、化學(xué)品工作者提供專業(yè)的化學(xué)品平臺(tái)進(jìn)行交流。
數(shù)據(jù)庫(kù)采用全中文化服務(wù),完全突破了中英文在化學(xué)物質(zhì)命名、化學(xué)品俗名、學(xué)名等方面的差異,所提供的數(shù)據(jù)全部中文化,更方便國(guó)內(nèi)從事化學(xué)、化工、材料、生物、環(huán)境等化學(xué)相關(guān)行業(yè)的工作人員查詢使用。
關(guān)注我們
-
微信賬號(hào):物競(jìng)化學(xué)品數(shù)據(jù)庫(kù)

-
微博賬號(hào):wjhxp
聯(lián)系我們
上海市延長(zhǎng)路149號(hào)上海大學(xué)科技園412室
公司總機(jī): 021-56389801
訂購(gòu)電話: 4007001514
傳真電話: 021-56389802
客服電話: 021-56332350
電子郵件: wingch@basechem.org
Copyright ? 物競(jìng)數(shù)據(jù)庫(kù) 2009-2026.All Rights Reserved. 關(guān)于數(shù)據(jù)庫(kù) | 關(guān)于物競(jìng) | 法律聲明 | 熱門關(guān)鍵字
 滬公網(wǎng)安備 31010602001115號(hào)
滬公網(wǎng)安備 31010602001115號(hào)