碘資源的利用—高價有機碘的合成和反應
- 2012-04-19
- 專題
目前我們的研究重點是低毒性的三價超價有機碘(λ3—有機碘),以及其在有機化學合成中的充分利用。
碘是大原子半徑的鹵族元素,具有易極化和低負電性的特點。八隅體理論指出碘能夠形成高價態有機碘是由于其易升高的價鍵所致。
典型的λ3—有機碘有亞酰碘苯(聚合物)1,碘苯二乙酸2,雙(三氟乙酰氧基)碘苯3,羥基(對甲苯磺酰氧基)碘苯(科澤試劑)4,2—亞碘酰基苯甲酸5。它們作為氧化劑廣泛應用于活性亞甲基、雙鍵和三鍵、醇羥基和酚羥基、硫基和氨基化合物的氧化反應1)。包括上述常見的碘類物質,有20多種的λ3—有機碘化合物可直接從東京Kasei Kogyo公司購買。
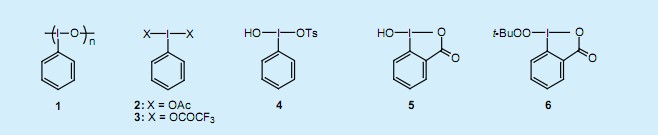
由于過氧烷基基團的λ3—有機碘化合物具有較強的易分解性,因此目前還不能成功合成。我們最近研究發現2)路易斯酸催化使1-羥基-1,2苯碘酰基-3(1氫)-酮 5和過氧叔丁酸進行配位體交換,得到過氧碘化物6晶體。過氧碘化物6在固體狀態下非常穩定,不易分解,而且它的結晶形態在室溫下可以較好地保存一年以上。這個產品引起人們的關注是因為它在同一個分子中含有過氧化叔丁基和一個三價的碘原子,而兩者都是極強的氧化劑。本文的目的是為了說明λ3—過氧碘化物的有用性及其作為自由基強氧化劑的廣泛應用。
1.烯基碘化物和炔基碘化物的合成與反應
在1985年人們發現亞碘酰苯1與BF3在二氯甲烷中活化,并與乙烯基硅烷或乙烯基錫烷反應,其中硅或者錫被三價的碘取代,生成λ3—烯基-碘化合物7并能取得較好產率(圖1)3)。λ3—烯基-碘化合物的生成是立體定向的,且乙烯基硅烷和乙烯基錫烷的立體化學結構保持不變。λ3—烯基-碘化合物7也可以通過以乙烯基硼酸為基質的碘化硼交換反應制得。
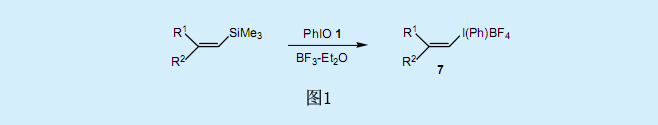
三價碘基團具有很強的基團離去性能,因此烯基碘化物7中乙烯基碳的親核取代反應在室溫的溫和條件下就能進行。在這些反應中,烯醇基陰離子、R2CuLi、CuCN、ArSNa、n-Bu4NC等可作為良好的親核試劑。特別的,伴隨著立體化學構型的轉化,發生了乙烯基碳的SN2(雙分子親核取代)反應;以前人們認為這個反應不能進行,而烯基碘化物7使這個反應成為了可能。碘苯還原消除反應后,烯基碘化物7中的α-氫原子具有足夠的酸性可用于α-消除反應的進行,且在一定基質存在的條件下,生成較好產率的亞烷基碳烯。
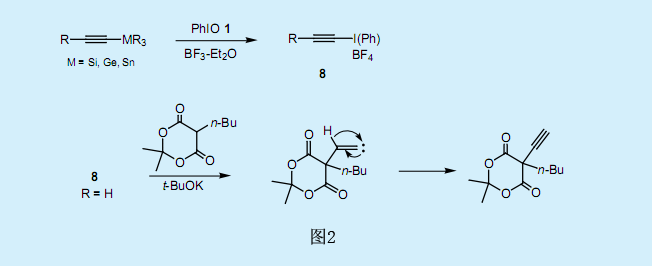
與烯基碘化物的合成方法類似,可以利用炔基硅烷和炔基錫烷合成炔基碘化物8(圖2)。炔基碘化物可直接從末端炔基開始進行合成5)。炔基碘化物8和烯醇基陰離子發生邁克爾加成反應形成環戊烯骨架。如圖2所示,炔基碘化物8(R=H)是一種良好的乙炔化試劑6)(可以向東京Kasei公司購買)。亞烷基碳烯是反應的中間產物,而炔化產物則可以通過將α-氫中的1,2號氫原子重排獲得。
2.丙二烯基碘化物的合成及其還原性碘-克萊森重排反應
有機硅酮-碘和炔丙基硅烷交換反應的應用催生了由SE2(雙分子親電取代反應)反應生成λ3—炔基碘化物。當碘苯二乙酸2與炔丙基硅烷9在BF3存在條件下反應時,炔丙基被接枝到碘苯分子的鄰位上,并生成了高產率的鄰炔丙基碘代苯107)(圖3)。在研究了若干其他反應之后,我們獲得了以下的觀察結果:
1)三價碘總是被還原為單加碘。
2)炔丙基化反應總是在碘基團的鄰位上發生。
3)新的C-C鍵會特定性地在丙炔基的α-碳上形成。
4)反應一個較低的溫度下進行。
5)丙炔基鍺烷和乙炔基錫烷也可用于反應。
6)羥基丙炔基硅烷和乙酸基丙炔基硅烷可用于反應。
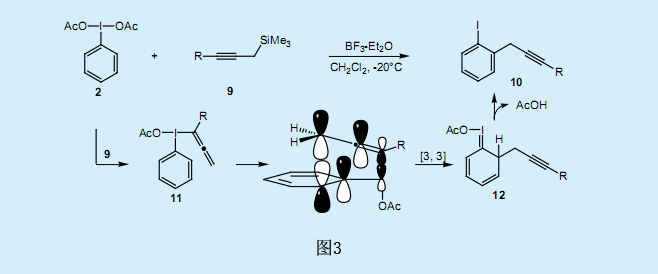
綜合以上的觀察結果,反應機理如圖3所示。炔丙基硅烷可與親電子試劑,例如鹵族元素和酰氯物質,發生SE2反應。首先,反應生成了λ3—丙二烯基碘化物11。隨后,經分子內的[3,3]單鍵轉移重排反應,炔丙基基團轉移至鄰位上,接著乙酸基隨著炔丙基碘代苯10的形成及12的還原消除反應而被釋放出來。λ3—丙二烯基碘化物11進行分子內重排時,其分子性質取決于單價碘烯烴化合物的交叉反應。盡管普通的克萊森重排反應需要加熱到150-250℃,但還原性碘-克萊森重排反應在低溫下就可以進行。假設反應的速率控控制步驟為λ3—丙二烯基碘化物11的[3,3]單鍵轉移重排反應。由于破壞頂點的碳-碘(Ⅲ)鍵只需要很小的能量,所以11的碘—克萊森重排反應所需的活化能較低。一般來說,芳香基-碘化物ArIX2具有T形幾何形狀,超價I(Ⅲ)—X鍵與芳香基的π鍵很好的相連。這種有利的軌道相互作用能夠促進11的重排反應發生。
在上述的還原性碘—克萊森重排反應中,由于沒有λ3—丙二烯基碘化物11生成的直接證據,我們可以認為它是一個中間產物。因此,為了分離和檢測中間產物—丙二烯基碘苯14,我們做了二甲基丙炔基硅烷13與亞碘酰基苯甲酸5的反應(圖4)。然而看似合理的假設—丙二烯基碘苯的[3,3]單鍵轉移反應卻較難進行,這可能由于分子末端的兩個甲基的位阻及其電子效應所導致的[3,3]單鍵轉移反應中超價碳—碘(Ⅲ)鍵斷裂卻沒有和芳香基的π軌道重疊造成的。和預期一樣,我們沒有在[3,3]單鍵轉移反應中發現鄰位的炔丙基化反應。遺憾的是,我們沒有檢測到丙二烯基碘苯14的生成。與我們的預期相反,該反應生成了過氧烷基碘化物15。過氧碘化物15在同一個分子中含有過氧烷基基團和超價碘(Ⅲ),其均可作為氧化劑,但是在固態時較穩定。X射線衍射分析顯示碘原子具有典型的T形幾何結構,且具有超價化合物形狀上局部變形的特點。
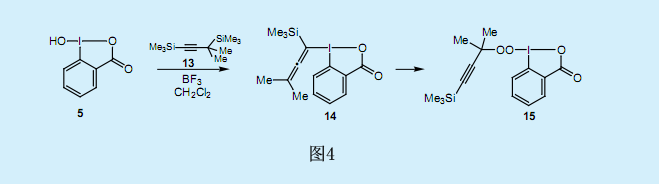
3.過氧化叔丁基碘化物的合成
超價λ3-有機碘化物具有一個作為配位體的過氧烷基基團,而這一結構是非常不穩定的。1968年Milas和Plesnecar報道稱,在-80℃的二氯甲烷中過氧叔丁酸可與亞碘酰苯1反應生成過氧化叔丁基自由基和碘苯。假設在該反應中,碘原子上的初始配體交換生成了不穩定的雙過氧烷基碘化物,從而導致O-I超價鍵的均勻斷裂,并生成了過氧化叔丁基自由基。由于過氧烷基碘化物15在室溫下非常穩定,因此其分離就十分地引人關注了。這可歸因于五元雜環化合物的形成所導致的同一位置上頂端雜環配體和中線芳香配體的固定作用。由于易斷裂的I-O鍵與苯基的π軌道間沒有的軌道的相互作用,因此這種排列所形成的亞酰碘苯甲基酮就使得過氧烷基碘化物15的穩定性增強。
這種具有獨特結構的過氧烷基碘化物預計可用來作為有機合成中的一種新型氧化劑。據此,設計引入過氧化叔丁基并嘗試合成碘化物6,以其作為典型化合物。亞酰碘基苯甲酸5在室溫下不與過氧叔丁酸發生反應,且由于其活性較差很容易被還原。然而,當反應混合物中加入路易斯酸后,碘原子上的配體交換反應按期望的進行,生成高產率過氧碘化物6(見圖5)。鄰亞酰碘基苯甲酸5的氧原子上的BF5配位作用使得反應活化。產物過氧化叔丁基碘化物6在固體狀態下是很穩定的,且在室溫下以晶體狀態保存一年以上也未見其分解。

盡管過氧碘化物6在固體狀態下是穩定的,但其在溶液中卻很容易分解。當過氧碘化物6在室溫下溶于氯仿中后,通過配位體交換反應就生成氯碘氧化物,其中過氧碘化物6的半衰期大約為4天。將結晶化的過氧碘化物在140℃下加熱,其迅速地分解為1,2-間二碘苯(46%)、碘苯(6%)、鄰碘苯甲酸(14%)和丙酮(43%)。在這一吸熱分解反應中,由于過氧基團與碘原子間弱超價鍵的斷裂,生成了過氧化叔丁基和9-碘-2σ-碘酰氧基16,進而導致了分解反應的發生。
4.經由過氧化叔丁基碘化物氧化的芐基氧化反應
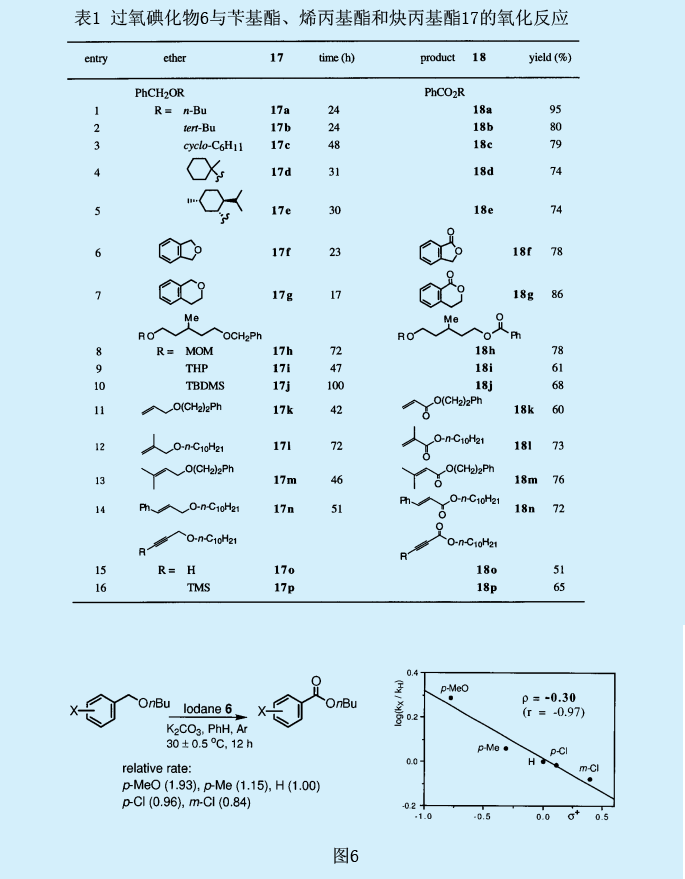
過氧化叔丁基碘化物6可有效的應用于芐基醚17上的芐基—亞甲基基團的氧化,生成苯甲酸酯類18。這些反應可在室溫下的氮氣環境中發生。酯類的產量與溶液的介電常數息息相關,盡管室溫下這一反應非常慢,但在具有較小介電常數的苯中還是得到了最佳的反應結果。不過,在苯中添加堿金屬碳酸鹽可以相當程度地加快這一反應。
在有機合成反應中,芐基經常被用來作為醇類的保護基團。由于酯類容易水解成醇類,過氧碘化物6就為芐基提供了一種氧化去保護方式。與去保護反應相關的一類常見問題是它們的化學選擇性。芐基位上的選擇性氧化甚至在MOM基團、甲硅烷基團、乙酰基團或者四氫吡喃基團存在條件下也會發生。烯丙基也可用來作為醇類的保護基團,而過氧碘化物6則對將烯丙醚類氧化為相應的αβ-不飽和酯類有益。另外,其它芳香烴類也很容易發生芐基氧化,像茚滿、四氫化萘、二氯化蒽和茐等都很容易被氧化。這些反應的部分結果如表1所示。
自由基抑制劑像α-生育酚和galvinoxyl可抑制芐基上亞甲基的氧化,這也意味著自由基種類的混亂。為了證實那些自由基產自于芐基位上,用與碳自由基反應迅速的2,2,6,6-四甲基哌啶-N-氧化物(TEMPO)來捕獲芐基自由基。我們研究了丁基芐醚17a的氧化反應的取代效果(圖6)。對位或間位上吸電子基氯的引入減緩了反應速率,而對位上甲基烷氧基或甲基的引入則加快了反應速率。Hammet相關系數是在相對反應速率和取代常數σ+間建立的,其中ρ=-0.30。這一ρ值可與由苯甲酸基自由生成的二芐基醚中芐基上氫的分離常數ρ=-0..65相比較。氘的主要同位素效應檢測結果顯示為一個很大的值(KH/KD=12-14)。這一同位素效應也強有力地表明芐基中C-H鍵的斷裂為速率控制步驟。
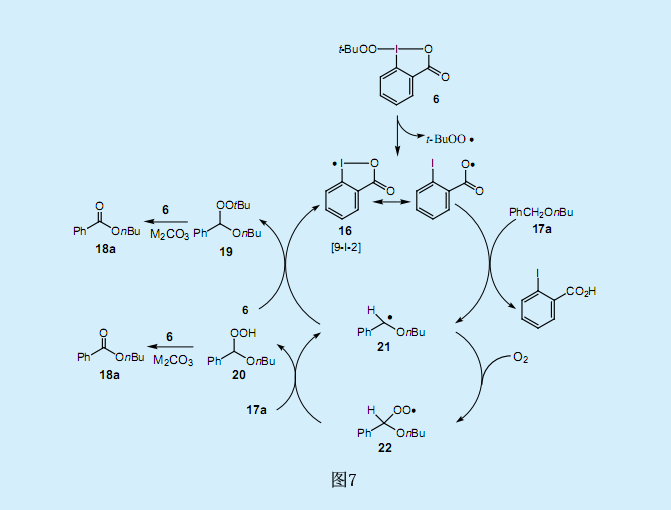
我們也對分子氧對反應的影響作了研究。十分引人關注的是,在過量丁基芐基醚17a(相對于過氧碘化物6而言)存在條件下,氮氣氛圍下的反應時間延長了達到了410h,并產生了比理論值更多的苯甲酸酯18a(約600%);而在無氧條件下,則生成了24%的苯甲酸酯18a和72%的過氧乙縮醛19(芐基位上含有過氧化叔丁基)。這一結果表明,反應的兩種中間產物過氧乙縮醛19和過氧乙羧酸20是由與分子氧的反應產生的。假設兩種中間產物均轉化為苯甲酸酯,反應機理如圖7所示。
首先,過氧基團的氧原子和過氧碘化物6中碘原子間的弱超價鍵斷裂,生成了過氧化叔丁基自由基和9-碘-2σ-碘酰氧基自由基16。當親電子的碘酰氧基自由基16吸取芐基醚17a的芐基上的氫就生成了芐基自由基21。芐基自由基21與過氧碘化物6進一步反應生成過氧化叔丁基乙縮醛19,并分解成相應的酯18a。另一方面,當反應系統中存在分子氧時,芐基自由基21與氧反應生成過氧基自由基22。過氧基自由基22與17a的芐基上的氫進一步反應生成過氧乙縮酸20和芐基自由基21。過氧乙縮酸20在一定反應條件下轉化為相應的酯18a。
5.經由過氧化叔丁基碘化物氧化的硫化物的氧化反應
過氧化叔丁基碘化物6將硫化物氧化為亞砜。乙腈溶液中二烴基硫化物和烷基芳香基硫化物可以高產率的轉化為亞砜(方法A)。二氯甲烷溶液中二烴基硫化物也可以高產率的轉化為亞砜(方法C)。在乙腈溶液中添加BF3-Et2O可以加快反應的進行(方法B)。
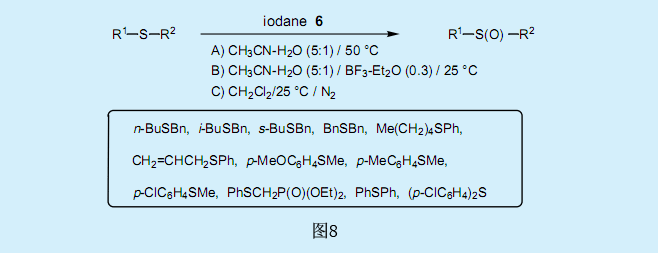
對乙腈溶液中苯甲硫醚取代反應的取代基效應研究表明,ρ對取代常數σ有一個很大的負值(-3.35)。而在乙腈溶液中添加BF3-Et2O后ρ值為-2.23,和取代常數σ+有更好的相關系數。從以下的觀察結果顯示,乙腈溶液中的反應為離子反應這一假定看來是合理的。
1)過氧碘化物6與亞酰碘苯甲酸5可達到共存平衡。
2)單一的以亞酰碘苯甲酸5或過氧化叔丁酸自身為氧化劑反應不能發生,但是當兩者共同使用時反應可以發生。
3)另外,添加自由基去除劑galvinoxyl對反應影響很小。
此外,這也充分地表明過氧化叔丁基碘化物6是活性物種,且在硫原子上產生了大量的帶有正電的活性中間體。另一方面,二氯甲烷(方法C)中添加galvinoxyl后完全抑制反應的發生表明,該反應的機理為自由基機理。
二硫縮醛的去保護氧化反應也會發生。將二硫縮醛與過氧碘化物6在乙腈中進行處理,發現它們能在數分鐘內完全反應并生成高產率的酮。過氧碘化物6可用來作為將硒醚氧化為硒亞砜和膦類氧化為膦氧化物的氧化劑。此外,過氧碘化物6也可以將2mol的三苯基膦氧化為相應的氧化物。
6.經由過氧化叔丁基碘化物氧化的胺類的氧化反應
過氧化叔丁基碘化物6可以有效的氧化胺類有機物。仲胺與過氧化叔丁基碘化物6發生脫水反應生成亞胺類有機物。碳酸鉀的添加可以加快反應的進行。四氫異喹啉氧化后可獲得高產率的二氫異喹啉。當使用過量的過氧碘化物6時則生成異喹啉。當過氧碘化物6與季胺類有機物反應時則生成過氧氨基縮醛,其中過氧基取代添加在胺的α碳原子上。

7.經由過氧化叔丁基碘化物氧化的酚類的自由基氧化反應
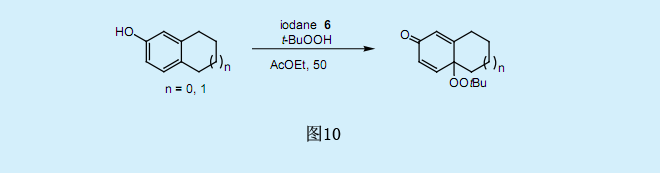
過氧化叔丁基碘化物與對位烷基取代的酚反應生成4-(過氧化叔丁基)環己二烯酮。在過氧化叔丁酸中,過氧碘化物6與對位取代的酚在溫和條件(乙酸乙酯/50。C)下反應生成高產率的過氧化叔丁基環己二烯酮。由于在galvinoxyl存在下氧化反應基本上被完全抑制了,而僅有少量副產物過氧化叔丁基環己二烯酮二聚物生成,因此我們假設這一反應為自由基反應。經由共振穩定的苯氧基自由基是一種反應中間體,它與過氧化叔丁基自由基耦合生成過氧化叔丁基環己二烯酮。
8.結論
在過氧化叔丁基碘化物6中,過氧化叔丁基與三價碘原子由超價鍵連接。大量關于其結構的調查研究使我們對其具有很高的活性很是期待,但其潛在的爆炸風險也令人擔憂。很可能僅有少數的化學家嘗試合成這種化合物。相反,令人驚訝的是結晶態的過氧化叔丁基碘化物6是十分穩定的,且在室溫下不會分解。另外,除非將其配制成溶液(但溶液中的反應也十分緩慢),否則組成超價鍵的自由基不會斷裂。一般來說,該反應在50℃以下才會進行,而超過這一溫度下的反應至今無人嘗試。
如上討論所言,過氧化叔丁基碘化物的開發是由我們集中力量尋求一種新的碘的潛在利用方式得到的。這些結果是由Takao Ito博士(日本煙草研究實驗室)精心實驗得到的。筆者在此衷心地感謝為此做出貢獻的學生們,他們的名字在本文所列的參考文獻中指出。
參考文獻:
1) a)G, F. Koser, in "The Chemistry of Functional Groups, Supplement D2"; Ed. by S. Patai and Z. Rappoport, Wiley, New York (1995); Chapters 21. b) A. Varvoglis, "The Organic Chemistry of Polycoordinated Iodine", VCH, New York (1992).
2) M. Ochiai, T. Ito, Y. Masaki, M. Shiro, J. Am. Chem. Soc., 114, 6269 (1992).
3) M. Ochiai, Kagaku Sosetsu, "Hypervalent Organic Compounds", 34, 181 (1998).
4) M. Ochiai, M. Toyonari, T. Nagaoka, D.-W. Chen, M. Kida, Tetrahedron Lett., 38, 6709 (1997).
5) M. Ochiai, in "Chemistry in Hypervalent Compounds"; Ed. by K. Akiba, Wiley-VCH, New York (1999); Chapters 12.
6) M. Ochiai, T. Ito, Y. Takaoka, Y. Masaki, M. Kunishima, S. Tani, Y. Nagao, J. Chem. Soc., Chem. Commun., 1990, 118.
7) M. Ochiai, T. Ito, Y. Takaoka, Y. Masaki, J. Am. Chem. Soc., 113, 1319 (1991).
8) M. Ochiai, T. Ito, M. Shiro, J. Chem. Soc., Chem. Commun., 1993, 218.
9) M. Ochiai, T. Ito, H. Takahashi, A. Nakanishi, M. Toyonari, T. Sueda, S. Goto, M. Shiro, J. Am. Chem. Soc., 118, 716 (1996).
10) M. Ochiai, A. Nakanishi, T. Ito, J. Org. Chem., 62, 4253 (1997).
11) M. Ochiai, D. Kajishima, T. Sueda, Heterocycles, 46, 71 (1997).
12) M. Ochiai, A. Nakanishi, A. Yamada, Tetrahedron Lett., 38, 3927 (1997).
注:本文為提供者翻譯的,由于知識所限,其中錯誤在所難免,敬請原諒。如有問題可以查找原文。
快速導航
化學品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
關于物競
物競數據庫是一個全面、專業、專注,并且免費的中文化學品信息庫,為學生、學者、化學品研究機構、檢測機構、化學品工作者提供專業的化學品平臺進行交流。
數據庫采用全中文化服務,完全突破了中英文在化學物質命名、化學品俗名、學名等方面的差異,所提供的數據全部中文化,更方便國內從事化學、化工、材料、生物、環境等化學相關行業的工作人員查詢使用。
關注我們

-
微信賬號:物競化學品數據庫

-
微博賬號:wjhxp
聯系我們
上海市延長路149號上海大學科技園412室
公司總機: 021-56389801
訂購電話: 4007001514
傳真電話: 021-56389802
客服電話: 021-56332350
電子郵件: wingch@basechem.org
 滬公網安備 31010602001115號
滬公網安備 31010602001115號