含較重主族元素的多重鍵化合物的新化學(xué)進(jìn)展
- 2012-04-18
- 專題
1. 引言
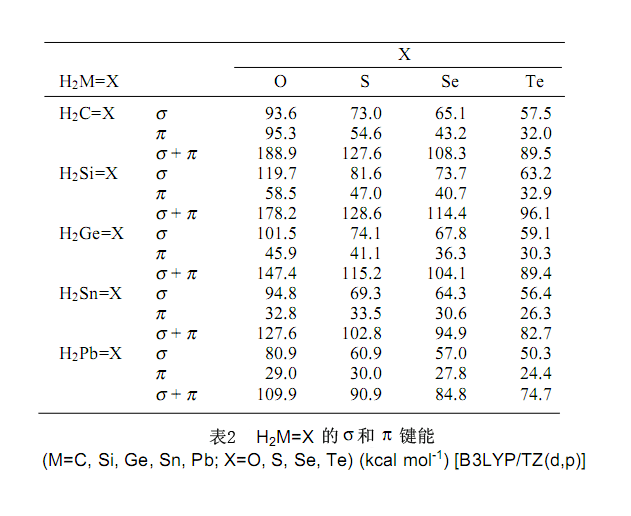
像烯烴、亞胺(希夫堿)、酮、乙炔、腈等含有第二周期元素的多重鍵化合物是穩(wěn)定的化合物,并且在有機(jī)化學(xué)中扮演一個(gè)很重要的角色。與此相對(duì)的是,含有來自第三周期前面的元素(被稱為較重的主族元素)的多鍵化合物有長(zhǎng)鍵距,而且通過P-軌道重迭產(chǎn)生的π鍵能很小所以它們極不穩(wěn)定。在表1中列舉了π鍵能的一些例子。
因?yàn)檫@種不穩(wěn)定性,直到1960年,所謂的“雙鍵規(guī)則”在課本被描述為“含有來自第三周期前面的元素的穩(wěn)定的雙鍵是不存在的。”
然而,證實(shí)這些化合物存在于低溫基質(zhì)或氣相中的證據(jù)已經(jīng)從研究累積起來,這些研究開始于1960s后半葉到1970s之間。第一次合成并分離出含有P=C鍵3和Si=C4、Si=Si5、P=P6的穩(wěn)定化合物分別在1978年和1981年。從那時(shí)起,關(guān)于與較重的主族元素相關(guān)的多重鍵化合物的化學(xué)發(fā)展迅速,并且?guī)缀醭蔀橹髯逶鼗瘜W(xué)中的領(lǐng)導(dǎo)主題。在本文,我們介紹這個(gè)領(lǐng)域的最新進(jìn)展,重點(diǎn)放在作者的研究上。
2. 含14族元素(硅,鍺,錫,鉛)的雙鍵化合物
2.1 較重主族元素的烯烴類似物
2.1.1 雙硅(Si-Si雙鍵化合物)
以乙烯為代表的C-C雙鍵化合物(烯烴)在有機(jī)化學(xué)中扮演著一個(gè)重要的角色。盡管烯烴有中等的反應(yīng)活性,但烯烴通常是一個(gè)穩(wěn)定的化合物。因此,很自然地,我們想嘗試合成這種含有硅——同屬于14族但位于碳下方——的雙鍵化合物。然而,自從二十世紀(jì)末以來,這種實(shí)驗(yàn)結(jié)果失敗了。獲得的產(chǎn)品已經(jīng)證實(shí)是它的低聚物或聚合物雙鍵,而非雙鍵化合物。在這種背景下,基于這些實(shí)驗(yàn),前面提到的“雙鍵規(guī)則”就成立了。已經(jīng)被證實(shí)的是,含雙鍵的碳和硅上位需取代基的引入作為一種防止聚合的手段是可行的。鑒于它通過降低與不穩(wěn)定化學(xué)物質(zhì)相關(guān)的反應(yīng)速率來穩(wěn)定,這種方法或?qū)⒈幻麨閯?dòng)力學(xué)穩(wěn)定,或者因?yàn)樗皿w積大的取代基保護(hù)高反應(yīng)活性的雙鍵而被命名為位保護(hù)。應(yīng)用該技術(shù),在1981年,Brook等4和West等5分別報(bào)道了含有第一個(gè)穩(wěn)定碳-硅雙鍵的硅烯1(AD=1-金剛烷基)和有一個(gè)硅-硅雙鍵的二硅烯2(Mes=苯基)。在West報(bào)道的不久后,Masamune等用一種不同的方法合成了二硅烯3。8隨后,又發(fā)展了幾種制備硅烯9和二硅烯7的合成方法,所以現(xiàn)在知道了大量這類穩(wěn)定的化合物。
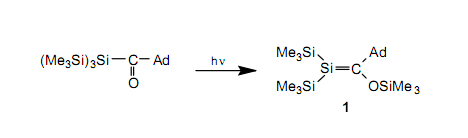
硅烯1,二硅烯2和二硅烯3在惰性的環(huán)境下是穩(wěn)定的,但與圖1中指出的烯烴相比,因?yàn)槎柘┯懈逪OMO和較低的LUMO,所以它們顯示了極高的反應(yīng)活性。相對(duì)于無色的烯烴,這是二硅烯是黃色的原因。
二硅烯2以方式4與氧氣反應(yīng)生成5。10進(jìn)一步地,它與水、醇、酸等經(jīng)歷副反應(yīng)后很容易生成6。11
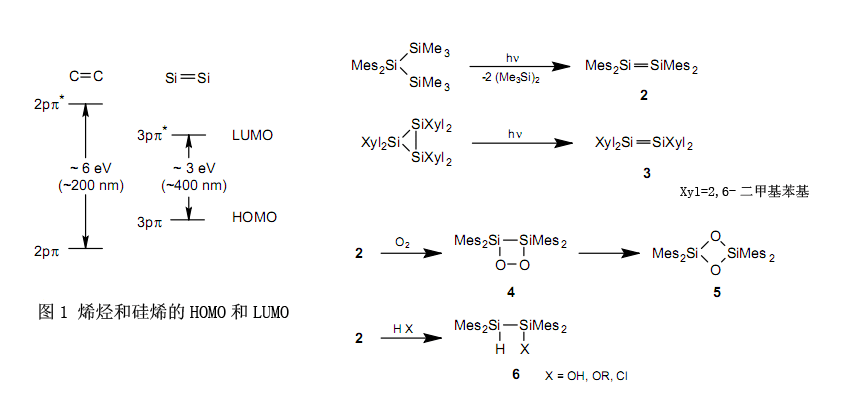
盡管事實(shí)上通過其他方法很難合成它們,但是利用二硅烯的高反應(yīng)活性可以合成多種含有新結(jié)構(gòu)的硅化合物。例如,通過與氧、硫、硒、碲、重氮甲烷、腈、疊氮化物等反應(yīng),以及與像苯乙酮這樣的酮類反應(yīng),可以很容易的分別合成下述含硅的三環(huán)化合物712和四環(huán)化合物8。13

二硅烯2和3與空氣接觸時(shí)立刻分解。通過添加苯基到硅上,可以增加它們的穩(wěn)定性。例如,由于大的金剛烷基和三異丙基苯基的存在,室溫下化合物9和10的半衰期接近一天。我們已經(jīng)能夠合成比較穩(wěn)定的化合物11,它室溫下的半衰期大約一個(gè)月,這使它能在空氣中使用。這種化合物有體積大的2,4,6-三[二(三甲基甲硅烷基)甲基]苯基基團(tuán)(Tbt)。Tbt基團(tuán)在鄰位有四個(gè)體積大的三甲基甲硅烷基基團(tuán)。然而,由于氫取代芐位,在1位有足夠的空間,并且在這個(gè)方位上它允許發(fā)生足夠的反應(yīng)以進(jìn)行功能基團(tuán)轉(zhuǎn)換。正如已被證實(shí)的,Tbt基團(tuán)對(duì)穩(wěn)定高周期的多鍵是非常有用的,這些多鍵通常有很高的反應(yīng)活性。
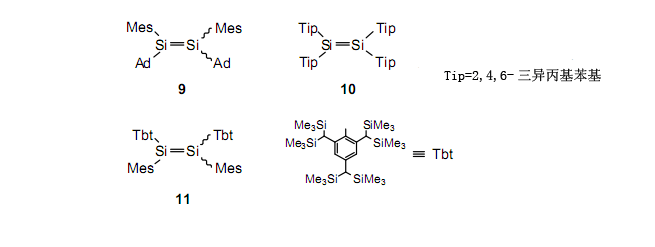
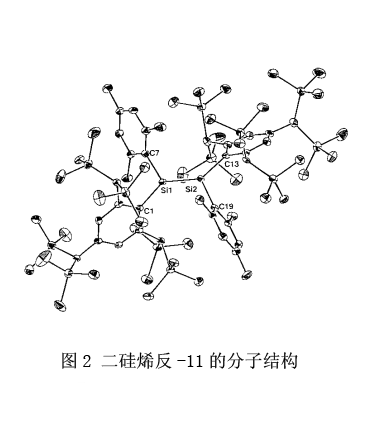
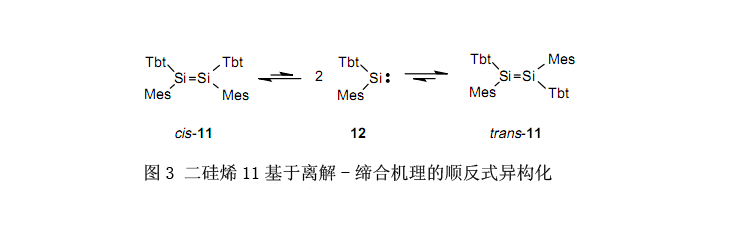
有趣的是,二硅烯11有極大的Tbt基團(tuán),所以能夠在三個(gè)方向上保護(hù)Si-Si雙鍵。另一方面,與其它二硅烯相比,硅-硅鍵被拉長(zhǎng)所以很容易分解成硅烯。16 表216列舉了二硅烯11(反式)的X射線晶體結(jié)構(gòu)分析。在含有大的芳基取代基的二硅烯中最長(zhǎng)的Si-Si鍵長(zhǎng)是2.228?。
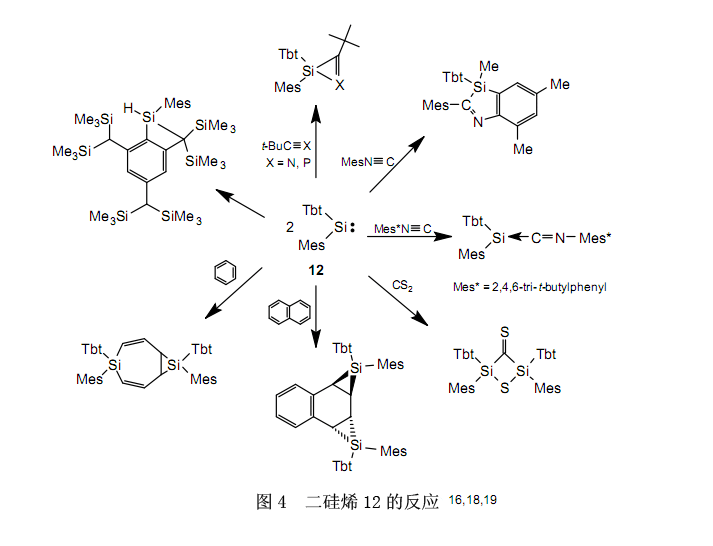
在溶液中,二硅烯11在室溫下逐步分解成硅烯,而在70℃下極快分解。因此,在室溫下經(jīng)由硅烯的分解,反式-二硅烯11異構(gòu)化為順式-二硅烯11和順式-二硅烯11異構(gòu)化為反式-二硅烯11分別進(jìn)行。眾所周知,烯烴的異構(gòu)化作用不是通過C-C鍵的裂解發(fā)生, 而是通過鍵的旋轉(zhuǎn)。而且,假設(shè)在硅上含有相對(duì)較小尺寸的取代基的已知二硅烯,異構(gòu)化作用與在烯烴上一樣通過Si-Si鍵的旋轉(zhuǎn)產(chǎn)生。二硅烯11是第一個(gè)基于離解-締合規(guī)則異構(gòu)化的二硅烯,了解這一點(diǎn)是很有趣的(圖3)。雖然至今為止通過熱解或光解作用來生產(chǎn)硅烯,但是在溫和的環(huán)境下產(chǎn)生硅烯方法的發(fā)現(xiàn)已經(jīng)使合成新的有機(jī)硅化合物成為可能,盡管事實(shí)上難以利用以前的方法合成它。圖4列舉了幾個(gè)例子。16,18,19
2.1.2 鍺、錫、鉛雙鍵化合物(二鍺烯、錫烯、鉛烯)
因?yàn)殛P(guān)于Ge=Ge、Sn=Sn、Pb=Pb的π鍵能相對(duì)于涉及Si=Si的π鍵能比較小,所以含有與硅屬于同一族的鍺、錫、鉛的雙鍵化合物(命名為二鍺烯、錫烯、鉛烯)也同樣比較不穩(wěn)定。另一方面,經(jīng)由談及的雙鍵分解產(chǎn)生的二價(jià)化學(xué)物種鍺烯、錫烯和鉛烯是非常穩(wěn)定的。因此,有適當(dāng)尺寸的取代基對(duì)于穩(wěn)定的雙鍵化合物是很重要的。
雖然在1970s,Lappert等在二硅烯2未合成時(shí)先合成了二鍺烯1321,22和錫烯1421,22,但是研究Ge=Ge和Sn=Sn的性質(zhì)是不可能的,因?yàn)樗麄円枣N烯15和stannylene16的形式分別存在于溶液中,盡管事實(shí)上它們作為雙鍵化合物存在于固相。二硅烯2和3合成之后,進(jìn)行了不引起分解而合成二鍺烯和錫烯的研究。結(jié)果是合成了化合物1723和1824。
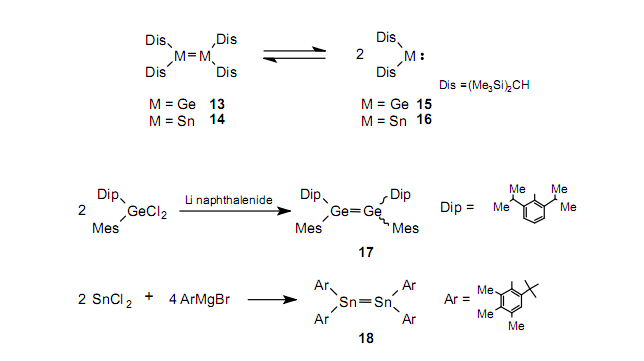
雖然二鍺烯17不分解成鍺烯,但是含有較大取代基的二鍺烯19易分解成鍺烯20,并在兩種物質(zhì)間存在一個(gè)平衡。25
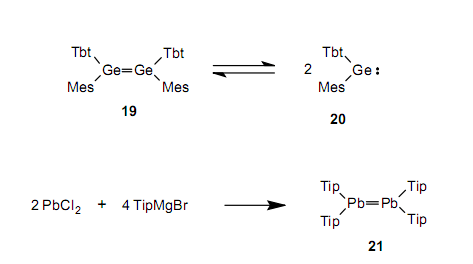
在1999年,也合成了含一個(gè)雙鍵的鉛烯21,這個(gè)雙鍵在占據(jù)周期表中最低位置的鉛原子之間。26因此,在west等合成二硅烯2后的18年里,已經(jīng)完成了屬于14族的相同元素之間的穩(wěn)定的雙鍵化合物。
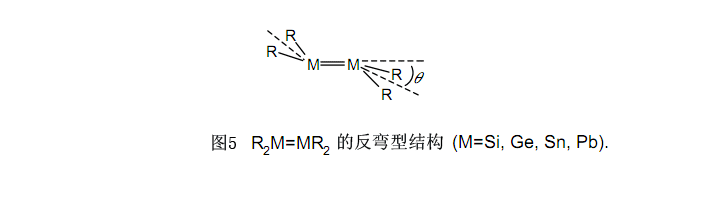
例如2,17-19,21的R2M=MR2化合物(M=Si,Ge,Sn,Pb)具有有趣的結(jié)構(gòu)特性,這是不同于烯烴的。這些化合物有一個(gè)所謂的“反彎型結(jié)構(gòu)”。7,20如在圖5中所列舉的,在一個(gè)M上的兩個(gè)取代基和另一個(gè)M上的兩個(gè)取代基以q被分彎曲成反式,而當(dāng)原子數(shù)增加時(shí)與平面形成的彎曲角將變大。雖然在本文未描述細(xì)節(jié),但是據(jù)透露,已經(jīng)通過R2M:的單線態(tài)和三線態(tài)間的能量差異確定了彎曲角,而且采取這樣一個(gè)與單線態(tài)穩(wěn)定程度相稱的彎曲結(jié)構(gòu)是比較恰當(dāng)?shù)摹?/p>
2.2 含硅的芳香族化合物
和烯烴一樣,含有不飽和鍵的主要有機(jī)化合物是包括苯,萘等在內(nèi)的芳香族化合物。因此,硅取代苯和萘中的碳得到的化合物是否展現(xiàn)出芳香族的性質(zhì),這個(gè)問題引起了極大的興趣。在1970s到1980s這個(gè)時(shí)期里進(jìn)行了大量的研究。在低溫基質(zhì)(例如,10K下的氬基質(zhì))或在氣相中,在硅上有像羥基,甲基基團(tuán)等簡(jiǎn)單取代基的硅雜苯22(R=H,Me)可以存在一小段時(shí)間,而且已經(jīng)測(cè)定了UV,IR光譜等。28與此同時(shí),在1988年M?rkl等報(bào)道的23已經(jīng)是一個(gè)適度穩(wěn)定的獨(dú)立的化合物,盡管事實(shí)上為分離作為一個(gè)穩(wěn)定化合物的硅雜苯已經(jīng)做了大量的實(shí)驗(yàn)。29雖然硅上的一個(gè)異丁基基團(tuán)和鄰位的兩個(gè)三甲基甲硅烷基基團(tuán)位保護(hù)了23,但是它只是在-100℃下的溶液中穩(wěn)定。此外,在后面會(huì)介紹,內(nèi)環(huán)硅通過溶液中四氫呋喃的協(xié)調(diào)確保穩(wěn)定。
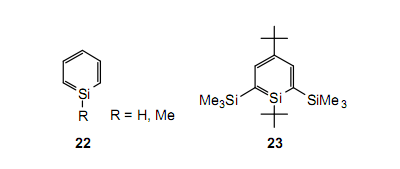
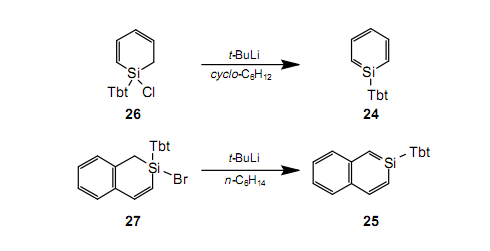
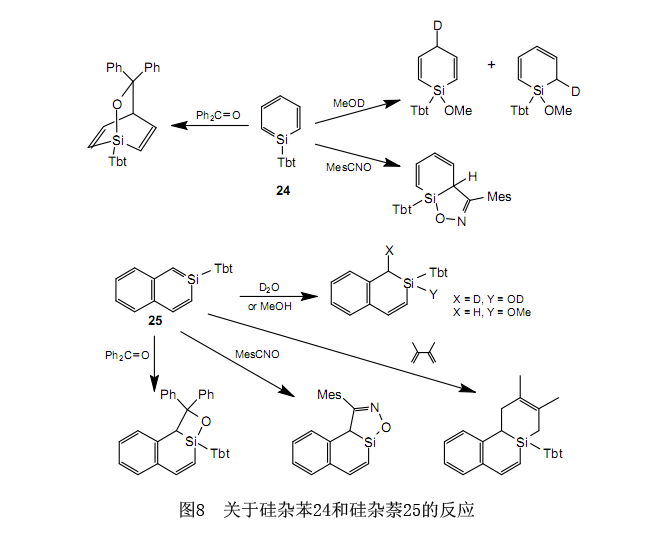
通過把Tbt基團(tuán)出色的保護(hù)能力應(yīng)用到含硅的芳香族化合物的合成上,我們已經(jīng)成功地分別分離出在室溫下作為穩(wěn)定晶體的硅雜苯2430和硅雜萘2531。24和25的合成前驅(qū)體分別是鹵化硅26和27。通過使用異丁基鋰時(shí)前驅(qū)體的脫氫鹵化作用獲得產(chǎn)品。
24和25都是無色的晶體,它們?cè)诙栊詺夥障路浅7€(wěn)定以至于在結(jié)晶態(tài)或溶液中完全不改變。29根據(jù)29Si的NMR,內(nèi)環(huán)硅的化學(xué)位移分別是87.3和92.5,這種情況存在于sp2硅的低磁場(chǎng)區(qū)域特性中。這完全不同于M?rkl等報(bào)道的化合物23的26.8的化學(xué)位移。在這種情況下,人們認(rèn)為作為溶劑的四氫呋喃明顯與23協(xié)調(diào),所以相應(yīng)地,它在較大程度上是穩(wěn)定的。表6列舉了內(nèi)環(huán)硅的耦合常數(shù)。兩個(gè)相鄰原子間的耦合常數(shù)與涉及鍵合的鍵級(jí)相聯(lián)系。依據(jù)圖6,硅雜苯24的耦合常數(shù)在硅雜萘25的兩個(gè)耦合常數(shù)之間,這恰好與苯和萘鍵級(jí)的關(guān)系是一致的。另外,一種典型的Si-C單鍵的耦合常數(shù)大約是50Hz。因此,24和25的Si-C鍵明顯具有了一個(gè)雙鍵的特征。結(jié)果是,π電子的離域作用明顯存在于24和25中,而且它們具有芳香族的特性。
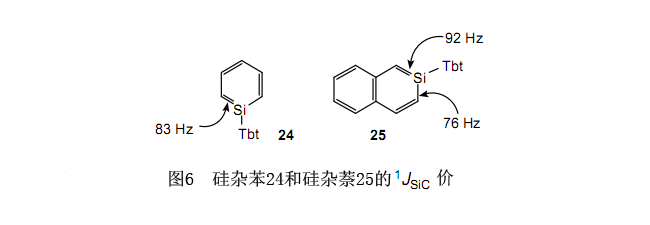
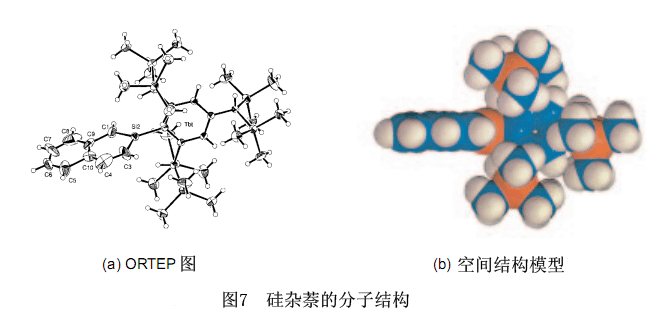
圖7顯示了25的X射線晶體結(jié)構(gòu)分析。硅雜苯的環(huán)狀部分位于平面上,而內(nèi)環(huán)硅和結(jié)合在它上的三個(gè)原子位于同一平面上。進(jìn)一步地,高活性的內(nèi)環(huán)硅完全由Tbt基團(tuán)保護(hù),從空間結(jié)構(gòu)模型(b)中可以清楚的看到。
從根本上說,24和25的電子光譜彼此相似,盡管事實(shí)上它們轉(zhuǎn)移到比相應(yīng)的苯和萘更長(zhǎng)的波長(zhǎng)。這也支持了24和25有芳香族特性這種情況。同時(shí),芳香族特性的量子計(jì)算理論變得更為可靠。Schleyer等建議NICS(核獨(dú)立化學(xué)位移)是一個(gè)關(guān)于芳香族特性的良好的指標(biāo)。32作為涉及硅雜苯與1-硅雜萘,2-硅雜萘和9-硅雜萘進(jìn)行的NICS計(jì)算的結(jié)果,他們的芳香族特性幾乎相同,盡管事實(shí)上它們比苯和萘的稍低,而且更進(jìn)一步,關(guān)于芳香族特性,硅雜萘的三種異構(gòu)體之間幾乎沒有差別。31計(jì)算的結(jié)果與從NMR,UV,X射線晶體結(jié)構(gòu)分析等中得到的實(shí)驗(yàn)結(jié)果是一致的。24和25的活性是很高的,盡管事實(shí)上它們?cè)诠枭嫌畜w積大的取代基并有推測(cè)的芳香族特性。在圖8中列舉了它們反應(yīng)的幾個(gè)例子。我們推斷,這是以與Si=C雙鍵相關(guān)的高反應(yīng)活性超過了來自于牽涉到硅雜苯和硅雜萘芳香族特性的穩(wěn)定性為依據(jù)的。
3. 含15族元素的雙鍵化合物(磷,砷,銻和鉍)
含N-N雙鍵的偶氮化合物也是與烯烴一樣眾所周知的化合物。類似地,在合成出硅烯1和二硅烯26同一年的1981年,yoshifuji等也第一次合成了含有一個(gè)穩(wěn)定P-P雙鍵的化合物——二磷烯28。
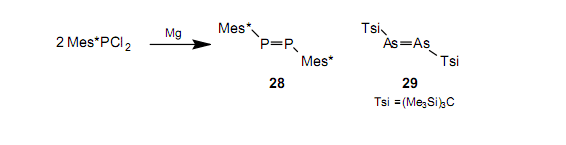
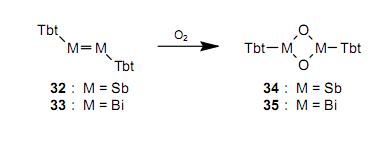
隨即,使用一個(gè)簡(jiǎn)單的方法也合成了一個(gè)含有As-As雙鍵的穩(wěn)定的化合物二砷烯。34由于這些雙鍵是逐漸不穩(wěn)定,所以盡管進(jìn)行了很多實(shí)驗(yàn),但是從未分別合成含有Sb-Sb和Bi-Bi之間雙鍵的化合物二銻烯和二鉍烯。人們期望,即使對(duì)15族中元素之間的雙鍵化合物, Tbt基團(tuán)也能夠提供位保護(hù)。在研究了很多合成方法之后,我們已經(jīng)第一時(shí)間分別合成并分離出作為綠色和紫色晶體的穩(wěn)定的二銻烯3235和二鉍烯3336。我們發(fā)明了一種新的合成方法,它通過使用三價(jià)磷試劑從六元化合物30和31上除去硒。
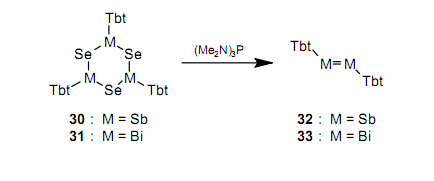
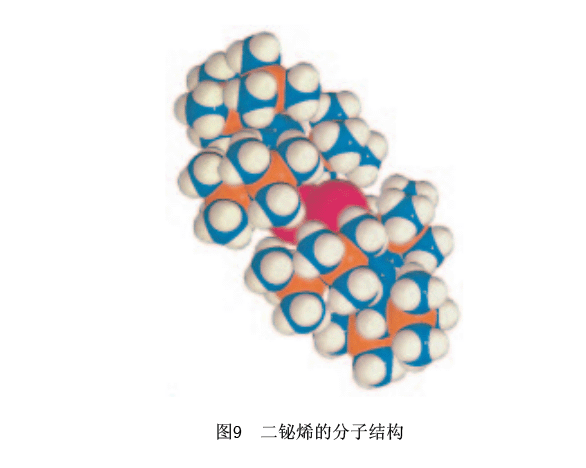
在有穩(wěn)定同位素的元素中,鉍是有最大的原子序數(shù)的第六周期元素。關(guān)于二鉍烯33的合成,人們的濃厚興趣不僅來自即使第六周期元素都可能產(chǎn)生雙鍵這一觀點(diǎn)(如前所述,在33合成之后,也合成了作為一個(gè)在其他的第六周期元素之間的雙鍵化合物二鉛烯21),而且也是來自它是有最大原子量的元素的雙鍵化合物的合成。通過X射線晶體結(jié)構(gòu)分析確定了與32和33有關(guān)的結(jié)構(gòu)。圖9列舉了33的分子結(jié)構(gòu),36從這里我們能夠推斷出體積大的Tbt基團(tuán)有效地保護(hù)了有高反應(yīng)活性的Bi-Bi雙鍵。
處于晶體態(tài)的32和33 是相當(dāng)穩(wěn)定的化合物,所以在空氣中,我們可以在短期內(nèi)處理它們。然而在空氣中,它們逐步與氧氣反應(yīng)產(chǎn)生四元環(huán)化合物34和35。非常有趣的是反應(yīng)通過維持晶相進(jìn)行,并且產(chǎn)生了單晶體34和35。35,36通過使用快速X射線衍射設(shè)備追蹤32的反應(yīng),可以發(fā)現(xiàn)反應(yīng)在大約20小時(shí)的誘導(dǎo)期之后突然開始。據(jù)推測(cè),在氧氣達(dá)到了一個(gè)臨界濃度后,氧氣從晶體表面進(jìn)入32,而且氧氣和32之間的反應(yīng)在一個(gè)像倒下的多米諾骨牌的拉伸下進(jìn)行。非常有趣的是當(dāng)維持單一晶相時(shí)沒有這種來自外部的反應(yīng)物反應(yīng)的例子。
4. 含16族元素(硫、硒、碲)的雙鍵化合物
不夸張地說,在有機(jī)化學(xué),特別是有機(jī)合成化學(xué)中,由第二周期元素產(chǎn)生的最重要的雙鍵化合物是以醛,酮等為代表的羰基化合物。研究一種正在討論中,以高周期元素取代羰基中氧原子的作用是極為有趣的。
4.1 碲酮
雖然很久之前已經(jīng)知道硫取代氧的硫酮(R2C=S),但是硒酮(R2C=S)是在1970s中期被Barton等合成出來。37Barton等并不能合成碲酮(R2C=Te)。
我們已經(jīng)開發(fā)出腙和二氯二硫38或二氯二硒39的反應(yīng),作為一種硫烯和硒烯的新合成方法。

通過把上述的反應(yīng)應(yīng)用到二氯化碲或四氯化碲,40a我們得到了含有雜環(huán)telluradiazoline36,通過熱解我們能夠由它合成第一個(gè)穩(wěn)定的碲酮37。40b

4.2 酮的較重元素類似物(重酮)
如果由來自14族較高周期的元素,例如硅、鍺、錫或鉛,取代酮中碳原子,產(chǎn)生的化合物就是我們指的當(dāng)作重酮的酮的較重元素類似物。此外,如果由來自16族的其他元素,例如硫、硒或碲取代氧原子,得到的化合物也會(huì)被指為重酮。
就像表2指出的,碳氧雙鍵的σ和π鍵能大約相當(dāng)。另一方面,所有的重酮中,σ鍵是強(qiáng)于π鍵的。因此,當(dāng)酮易于進(jìn)行加成和消去反應(yīng)時(shí),重酮更易于進(jìn)行由兩個(gè)更積極穩(wěn)定的σ鍵取代一個(gè)π鍵的加成反應(yīng)。由于這些積極地原因,合成重酮是很困難的。我們已經(jīng)能夠合成重酮化合物,這種物質(zhì)中體積大的取代基減少了π鍵的反應(yīng)活性。
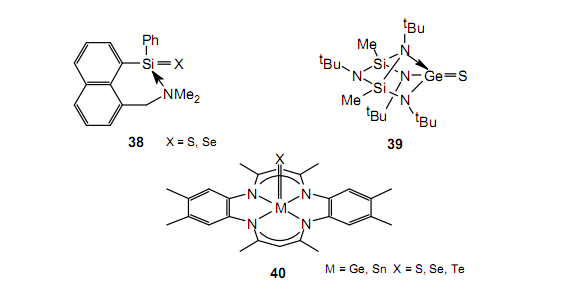
為了合成與重酮相關(guān)的雙重鍵化合物已經(jīng)進(jìn)行了很多合成實(shí)驗(yàn)。在化合物38-40中已經(jīng)列舉了幾個(gè)例子。其中一些并不是真的雙鍵化合物,因?yàn)樵谀撤N程度上通過雜原子的配位穩(wěn)定了雙鍵的特征。
這一次,我們也是通過使用高位保護(hù)能力的Tbt基團(tuán)才能合成許多穩(wěn)定的重酮。圖10列舉了兩種使用的合成方法。總之,方法(1)比較常見,但是當(dāng)作為開始原料的1,2,3,4-tetrachalcogenametalloranes的合成不是那么容易時(shí),方法(2)將會(huì)有用。

當(dāng)Tbt和Tip基團(tuán)共同存在于14族元素上時(shí),把硅烷硫酮4142、鍺烷硫酮4246,47鍺烷硒酮4347,48當(dāng)作穩(wěn)定的化合物分別分離出來是可以實(shí)現(xiàn)的。
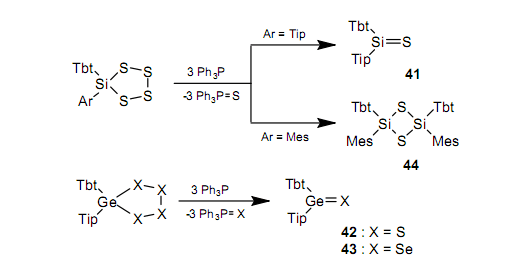
然而,當(dāng)一個(gè)較小的Mes基團(tuán)取代了Tip時(shí),分離的化合物是一個(gè)二聚物44,雖然我們能夠確認(rèn)通過被困反應(yīng)產(chǎn)生了作為中間體的Tbt(Mes)Si=S。所以為了分離重酮,具有位保護(hù)的體積大的取代基是很重要的。42依據(jù)Tbt和Tip取代基化合的方法不足以合成含錫的重酮。這是由于當(dāng)與含錫雙鍵相關(guān)的能量降低時(shí)提升了反應(yīng)活性,而Sn周圍的雙鍵變得更長(zhǎng)時(shí),位保護(hù)作用未有效地發(fā)揮作用。然而,在一個(gè)穩(wěn)定相中,憑借把Tip改為一個(gè)較大的取代基Ditp基團(tuán),我們也能夠分離錫烷硫酮4549和錫烷硒酮4650。
通過使用被困反應(yīng),在較低溫度下,能夠在脫硫作用中確認(rèn)鉛烷硫酮47,但是不可能分離出47。
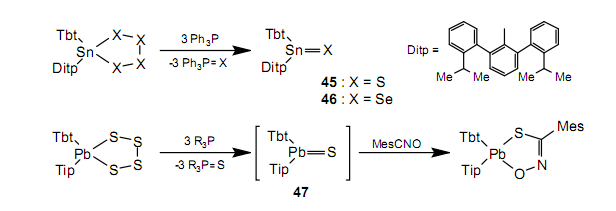
我們不能合成出相應(yīng)的tetraselenasilorane(R1R2SiSe4),所以我們無法通過脫硒化合成silaneselone(R1R2Si=Se)。然而通過使用silacyclopropane48作為硅烯49的先驅(qū)體的硒化反應(yīng),我們可以合成silaneselone50。52
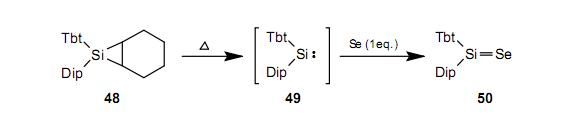
至于鍺,鍺環(huán)丙烯51,52起到作鍺烯 53,5453良好的先驅(qū)體。通過在硫和硒存在時(shí)加熱52,我們能夠獲得鍺烷硫酮55和鍺烷硒酮56。47對(duì)于Ge-Te雙鍵合成,這個(gè)反應(yīng)是特別有效的,而且我們能夠合出鍺烷碲酮57,58。54
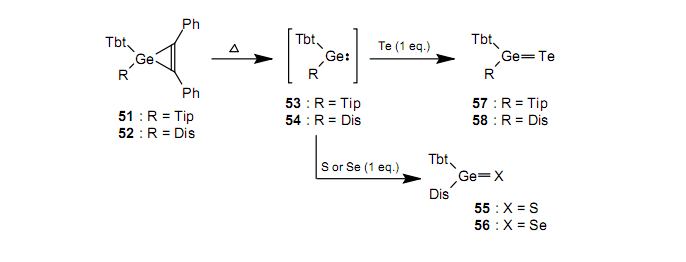
當(dāng)我們把分離出的鉛烯59與相當(dāng)量的硫反應(yīng)時(shí),我們得到了一種二價(jià)的化學(xué)物種鉛烯61。55
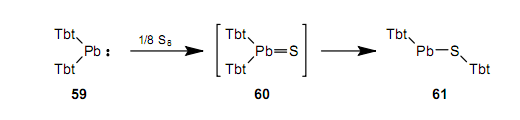
因此,就鉛而言,2-配位型61比3-配位型60更穩(wěn)定。這與量子化學(xué)計(jì)算的結(jié)果是一致的,結(jié)果顯示HPb(SH)比H2Pb=S穩(wěn)定了大約39kcal/mol。41a有關(guān)鉛的重酮有不同于輕類似物的性質(zhì)。
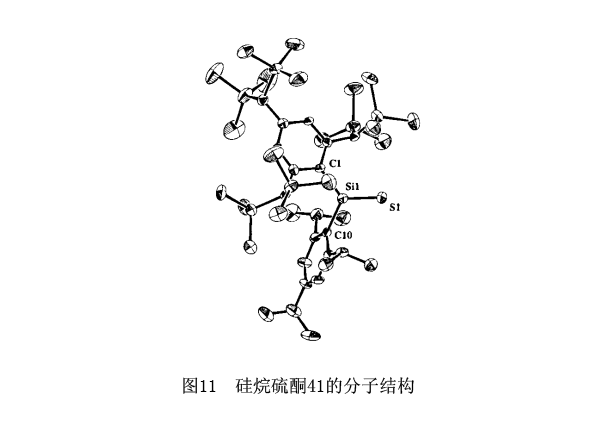
所有合成的重酮被觀察成有色晶體(41:黃色;42,55,黃橙色;43,45,46,50,56:紅色;57,58,綠色)。X射線晶體結(jié)構(gòu)分析確定了硅烷硫酮41,鍺烷硫酮42,鍺烷硒酮43,56,錫烷硒酮46和鍺烷碲酮57,58的結(jié)構(gòu)。圖11列舉了硅烷硫酮41的分子結(jié)構(gòu)。42
值得關(guān)注的結(jié)構(gòu)特性是硅周圍全部三角平面協(xié)調(diào)的鍵角都是360°,而且Si-S鍵距是1.948 ?,這比普通的Si-Si單鍵短到大約9%,所以很明顯它有雙鍵的特征。對(duì)于酮,所有這些數(shù)據(jù)都是完全相同的結(jié)構(gòu)特性。已經(jīng)得到實(shí)驗(yàn)證實(shí)的是重酮與酮結(jié)構(gòu)上是相同的。X射線晶體結(jié)構(gòu)分析確定的其他重酮呈現(xiàn)一樣的結(jié)構(gòu)特性。
在碳-氧雙鍵的反應(yīng)中,關(guān)于羰基基團(tuán)的重要反應(yīng)特性是可逆性,如在與水和醇的反應(yīng)中所示的一樣。在討論中,基于加成-消去機(jī)理的反應(yīng)經(jīng)由一個(gè)四配位的中間物進(jìn)行。
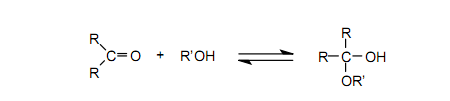
在表2中看到,與C=O鍵相關(guān),大約相等的σ鍵能和π鍵能使前面提到的方法成為可能。然而,假設(shè)重酮,加成反應(yīng)是顯著放熱并且不可逆,其原因很大程度上是π鍵能比σ鍵能低。事實(shí)上,所有合成的重酮迅速與水、醇等反應(yīng)并引發(fā)四配位副產(chǎn)物。另外,相對(duì)于酮的情況,重酮很容易和不飽和的化學(xué)物,例如苯基異硫氰酸鹽、mesitonitrile oxide、2,3-二甲基丁二烯等發(fā)生環(huán)加成反應(yīng)。42-50通過使用這些反應(yīng)我們能夠合成新的雜環(huán)化合物,盡管事實(shí)上通過一種其他方法難以合成它們。

5. 結(jié)論
通過使用體積大的取代基,在動(dòng)力學(xué)穩(wěn)定性基礎(chǔ)上合成了大量包含14,15和16族高周期元素的多重鍵化合物。更進(jìn)一步地,雖然我們?cè)诒疚闹袥]有涉及它們,但是也已經(jīng)合成了一些包含13族元素的多重鍵化合物。與高周期元素有關(guān)的多重鍵化合物與含有有趣結(jié)構(gòu)和反應(yīng)活性的獨(dú)特化合物一起已經(jīng)在有機(jī)化學(xué)和無機(jī)化學(xué)中建立了一個(gè)新的領(lǐng)域。然而,在這個(gè)領(lǐng)域中仍然留有大量正等待合成的化合物。例如,在14族元素之間的三鍵化合物,R-M≡M-R,含氧的重酮,R2M=O(M=Si,Ge,Sn和Pb)以及含Ge,Sn,Pb等的芳香族化合物。
參考文獻(xiàn):
1) M. W. Schmidt, P. N. Truong, M. S. Gordon, J. Am. Chem. Soc., 109, 5217 (1987); P. v. R. Schleyer, D. Kost,J. Am. Chem. Soc., 110, 2105 (1988).
2) K. S. Pitzer, J. Am. Chem. Soc., 70, 2140 (1948).
3) T. C. Klebach, R. Lourens, F. Bickelhaupt, J. Am. Chem. Soc., 100, 4886 (1978).
4) A. G. Brook, F. Abdesaken, B. Gutekunst, G. Gutekunst, R. K. Kallury, J. Chem. Soc., Chem. Commun.,1981, 191.
5) R. West, M. J. Fink, J. Michl, Science, 214, 1343 (1981).
6) M. Yoshifuji, I. Shima, N. Inamoto, K. Hirotsu, T. Higuchi, J. Am. Chem. Soc., 103, 4587 (1981).
7) Review: R. Okazaki, R. West, Adv. Organomet. Chem., 39, 233 (1996).
8) S. Masamune, Y. Hanzawa, S. Murakami, T. Bally, J. F. Blount, J. Am. Chem. Soc., 104, 1150 (1982).
9) Review: A. G. Brook, M. A. Brook, Adv. Organomet. Chem., 39, 71 (1996).
10) A. J. Millevolte, D. R. Powell, S. G. Johnson, R. West, Organometallics, 11, 1091 (1992); K. L. McKillop,G. R. Gillette, D. R. Powell, R. West, J. Am. Chem. Soc., 114, 5203 (1992); H. Sohn, R. P. Tan, D. R. Powell,R. West, Organometallics, 13, 1390 (1994).
11) D. N. Roark, G. J. D. Peddle, J. Am. Chem. Soc., 94, 5837 (1972); D. J. DeYoung, M. J. Fink, J. Michl,R. West, Main Group Met. Chem., 1, 19 (1987).
12) H. Piana, U. Schubert, J. Organomet. Chem., 348, C19 (1988); G. R. Gillette, R. West, J. Organomet. Chem.,394, 45 (1990); H. B. Yokelson, A. J. Millevolte, G. R. Gillette, R. West, J. Am. Chem. Soc., 109, 6865(1987); R. West, D. J. DeYoung, K. J. Haller, J. Am. Chem. Soc., 107, 4942 (1985); R. P. Tan, G. R. Gillette,D. R. Powell, R. West, Organometallics, 10, 546 (1991); H. B. Yokelson, A. J. Millevolte, K. J. Haller, R. West,J. Chem. Soc., Chem. Commun., 1987, 1605.
13) M. J. Fink, D. J. DeYoung, R. West, J, Michl, J. Am. Chem. Soc., 105, 1070 (1983); P. Boudjouk, B.-H. Han,K. R. Anderson, J. Am. Chem. Soc., 104, 4992 (1982); P. Boudjouk, Nachr. Chem., Tech. Lab., 31, 798(1983); A. D. Fanta, D. J. DeYoung, J. Belzner, R. West, Organometallics, 10, 3466 (1991).
14) B. D. Shepherd, D. R. Powell, R. West, Organometallics, 8, 2664 (1989).
15) R. S. Archibald, Y. van den Winkel, D. R. Powell, R. West, J. Organomet. Chem., 446, 67 (1993); H. Watanabe,K. Takeuchi, N. Fukawa, M. Kato, M. Goto, Y. Nagai, Chem. Lett., 1987, 1341.
16) N. Tokitoh, H. Suzuki, R. Okazaki, K. Ogawa, J. Am. Chem. Soc., 115, 10428 (1993); H. Suzuki, N. Tokitoh,R. Okazaki, J. Harada, K. Ogawa, S. Tomoda, M. Goto, Organometallics, 14, 1016 (1995); H. Suzuki,N. Tokitoh, R. Okazaki, Bull. Chem. Soc. Jpn., 68, 2471 (1995).
17) a) R. Okazaki, M. Unno, N. Inamoto, Chem. Lett., 1987, 2293. b) R. Okazaki, N. Tokitoh, T. Matsumoto,in “Synthetic Methods of Organometallic and Inorganic Chemistry,” ed by W. A. Herrmann, Thieme,New York (1996) Vol. 2, p. 260.
18) H. Suzuki, N. Tokitoh, R. Okazaki, J. Am. Chem. Soc., 116, 11572 (1994).
19) N. Tokitoh, H. Suzuki, R. Okazaki, J. Chem. Soc., Chem. Commun., 1996, 125; N. Takeda, H. Suzuki,N. Tokitoh, R. Okazaki, J. Am. Chem. Soc., 119, 1456 (1997).
20) Review: K. Baines, W. G. Stibbs, Adv. Oranomet. Chem., 39, 275 (1996).
21) P. J. Davidson, D. H. Harris, M. F. Lappert, J. Chem. Soc., Dalton Trans., 1976, 2268.
22) D. E. Goldberg, P. B. Hitchcock, M. F. Lappert, J. Chem. Soc., Dalton Trans., 1986, 2387.
23) S. A. Batcheller, T. Tsumuraya, O. Tempkin, W. M. Davis, S. Masamune, J. Am. Chem. Soc., 112, 9394(1990).
24) M. Weidenbruch, H. Killian, K. Peters, H. G. v. Schnering, Chem. Ber., 128, 983 (1995).
25) K. Kishikawa, N. Tokitoh, R. Okazaki, Chem. Lett., 1998, 239.
26) M. Stürmann, W. Saak, H. Marsmann, M. Weidenbruch, Angew. Chem., Int. Ed., 38, 187 (1999).
27) Y. Apeloig, M. Karni, in “The Chemistry of Organic Silicon Compounds,” ed by Z. Rappoport, Y. Apeloig,Wiley, New York (1998) Vol. 2, Chapter 1.
28) T. J. Barton, D. Banasink, J. Am. Chem. Soc., 99, 5199 (1977); H. Bock, R. A, Bowling, B. Solouki,T. J. Barton, G. T. Burns, J. Am. Chem. Soc., 102, 429 (1980); T. J. Barton, G. T. Burns, J. Am. Chem. Soc.,100, 5246 (1978); G. Maier, G. Mihm, H. P. Reisenauer, Angew. Chem., Int. Ed. Engl., 19, 52 (1980).
29) G. M?rkl, W. Schlosser, Angew. Chem., Int. Ed. Engl., 27, 963 (1988).
30) K. Wakita, N. Tokitoh, R. Okazaki, S. Nagase, Angew. Chem., Int. Ed., 39, 634 (2000).
31) N. Tokitoh, K. Wakita, R. Okazaki, S. Nagase, P. v. R. Schleyer, H. Jiao, J. Am. Chem. Soc., 119, 6951 (1997);K. Wakita, N. Tokitoh, R. Okazaki, S. Nagase, P. v. R. Schleyer, H. Jiao, J. Am. Chem. Soc., 122, 5648 (2000).
32) P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao, N. J. R. v. E. Hommes, J. Am. Chem. Soc., 118, 6317(1996).
33) M. Yoshifuji, in “Multiple Bonds and Low Coordination in Phosphorus Chemistry,” ed by M. Regitz,O. J. Scherer, Thieme, New York (1990), Chapter 9.
34) A. Cowley, J. G. Lasck, N. C. Norman, M. Pakulski, J. Am. Chem. Soc., 105, 5506 (1983).
35) N. Tokitoh, Y. Arai, T. Sasamori, R. Okazaki, S. Nagase, H. Uekusa, Y. Ohashi, J. Am. Chem. Soc., 120, 433(1998).
36) N. Tokitoh, Y. Arai, R. Okazaki, S. Nagase, Science, 277, 78 (1997).
37) T. G. Back, D. H. R. Barton, M. R. Britten-Kelly, F. S. Guziec, Jr., J. Chem. Soc., Perkin Trans. 1, 1976, 2079.
38) R. Okazaki, K. Inoue, N. Inamoto, Bull. Chem. Soc. Jpn., 54, 3541 (1981).
39) A. Ishii, R. Okazaki, N. Inamoto, Bull. Chem. Soc. Jpn., 61, 861 (1988).
40) a) M. Minoura, T. Kawashima, N. Tokitoh, R. Okazaki, Tetrahedron, 53, 8137 (1997). b) M. Minoura,T. Kawashima, R. Okazaki, J. Am. Chem. Soc., 115, 7019 (1993); M. Minoura, T. Kawashima, R. Okazaki,Tetrahedron Lett., 38, 2501 (1997).
41) Reviews: a) N. Kano, N. Tokitoh, R. Okazaki, Yuki Gosei Kagaku Kyokai Shi (J. Synth. Org. Chem. Japan),56, 919 (1998). b) N. Tokitoh, T. Matsumoto, R. Okazaki, Bull. Chem. Soc. Jpn., 72, 1665 (1999). c) N. Tokitoh, R. Okazaki, in “ The Chemistry of Organic Silicon Compounds,” ed by Z. Rappoport,Y. Apeloig, Wiley, New York (1998) Vol. 2, Chapter 17.
42) H. Suzuki, N. Tokitoh, R. Okazaki, S. Ngase, M. Goto, J. Am. Chem. Soc., 120, 11096 (1998).
43) R. Arya, J. Boyer, F. Carré, R. Corriu, G. Lanneau, J. Lapasset, M. Perrot, C. Priou, Angew. Chem., Int. Ed,Engl., 28, 1016 (1989).
44) a) M. Veith, S. Becker, V. Huch, Angew. Chem., Int. Ed, Engl., 28, 1237 (1989). b) M. Veith, A. Detemple,V. Huch, Chem. Ber., 124, 1135 (1991). c) M. Veith, A. Detemple, Phosphorus, Sulfur, Silicon, 65, 17 (1992).
45) a) M. C. Kuchta, G. Parkin, J. Chem. Soc., Chem. Commun., 1994, 1351. b) M. C. Kuchta, G. Parkin,J. Am. Chem. Soc., 116, 8372 (1994). c) W.-P. Leung, W.-H. Kwok, L. T. C. Law, Z.-Y. Zhou, T. C. W. Mak,J. Chem. Soc., Chem. Commun., 1996, 505.
46) N. Tokitoh, T. Matsumoto, K. Manmaru, R. Okazaki, J. Am. Chem. Soc., 115, 8855 (1993).
47) T. Matsumoto, N. Tokitoh, R. Okazaki, J. Am. Chem. Soc., 121, 8811 (1999).
48) T. Matsumoto, N. Tokitoh, R. Okazaki, Angew. Chem., Int. Ed. Engl., 33, 2316 (1994).
49) M. Saito, Ph.D. Thesis, The University of Tokyo, 1996.
50) M. Saito, N. Tokitoh, R. Okazaki, J. Am. Chem. Soc., 119, 11124 (1997).
51) N. Kano, N. Tokitoh, R. Okazaki, Chem. Lett., 1997, 277; N. Kano, N. Tokitoh, R. Okazaki, Organometallics,16, 4237 (1997).
52) T. Sadahiro, N. Tokitoh, R. Okazaki, unpublished results.
53) N. Tokitoh, K. Kishikawa, T. Matsumoto, R. Okazaki, Chem. Lett., 1995, 827.
54) N. Tokitoh, T. Matsumoto, R. Okazaki, J. Am. Chem. Soc., 119, 2337 (1997).
55) N. Kano, Ph.D. Thesis, The University of Tokyo, 1998.
注:本文為提供者翻譯的,由于知識(shí)所限,其中錯(cuò)誤在所難免,敬請(qǐng)?jiān)彙H缬袉栴}可以查找原文。
統(tǒng)計(jì)數(shù)據(jù)
共收錄化學(xué)品數(shù)據(jù)
147579 條
公告
更多>
-
微信掃一掃
關(guān)注:物競(jìng)化學(xué)品數(shù)據(jù)庫(kù)
-
物竟數(shù)據(jù)庫(kù) 手機(jī)版
國(guó)內(nèi)首款化工類專業(yè)手機(jī)應(yīng)用
-
微博賬號(hào)
wjhxp
快速導(dǎo)航
化學(xué)品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
關(guān)于物競(jìng)
物競(jìng)數(shù)據(jù)庫(kù)是一個(gè)全面、專業(yè)、專注,并且免費(fèi)的中文化學(xué)品信息庫(kù),為學(xué)生、學(xué)者、化學(xué)品研究機(jī)構(gòu)、檢測(cè)機(jī)構(gòu)、化學(xué)品工作者提供專業(yè)的化學(xué)品平臺(tái)進(jìn)行交流。
數(shù)據(jù)庫(kù)采用全中文化服務(wù),完全突破了中英文在化學(xué)物質(zhì)命名、化學(xué)品俗名、學(xué)名等方面的差異,所提供的數(shù)據(jù)全部中文化,更方便國(guó)內(nèi)從事化學(xué)、化工、材料、生物、環(huán)境等化學(xué)相關(guān)行業(yè)的工作人員查詢使用。
關(guān)注我們
-
微信賬號(hào):物競(jìng)化學(xué)品數(shù)據(jù)庫(kù)

-
微博賬號(hào):wjhxp
聯(lián)系我們
上海市延長(zhǎng)路149號(hào)上海大學(xué)科技園412室
公司總機(jī): 021-56389801
訂購(gòu)電話: 4007001514
傳真電話: 021-56389802
客服電話: 021-56332350
電子郵件: wingch@basechem.org
Copyright ? 物競(jìng)數(shù)據(jù)庫(kù) 2009-2026.All Rights Reserved. 關(guān)于數(shù)據(jù)庫(kù) | 關(guān)于物競(jìng) | 法律聲明 | 熱門關(guān)鍵字
 滬公網(wǎng)安備 31010602001115號(hào)
滬公網(wǎng)安備 31010602001115號(hào)