利用橫向分子間相互作用控制液晶分子超晶格結構
- 2012-04-10
- 專題
1. 介紹
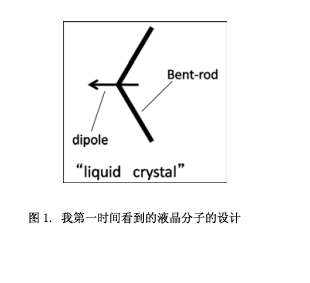
1997年的5月,那個時候我正在想“我應該怎樣來改變我的化學?”從學生時代我都一直在做有機化學的研究項目,而那個時候,我是一名35歲的助理教授.我無法停止思考這個問題,怎樣去擴展我的化學領域,我認為我必須得開始著手一個與我現在正在的研究方向完全不同的領域。然后我決定去美國或者是歐洲學習超分子化學或者是材料化學。然而, 那個時候我在美國或者是歐洲沒有有聯系的教授,因此我給在這些領域里面做得很好的專家發郵件,爭取一個博士后的學習機會。第一個為我提供機會的是麻省理工學院的Timothy M. Swager教授,他是超分子化學和材料科學領域的權威,幸運的是,他回復我“你將什么時間過來?”然后我開始了麻省理工學院的學習。在第一天,Swager教授給了我一張紙,上面寫了一個簡單的字符 (圖1),在橫向方向的彎曲桿的中心有一個箭頭。他說“如果你合成這類分子,就會發現有趣的現象”。這似乎是一個“液晶”的研究.隨后,我查閱了英日字典意識到這個“液晶”是一個存于晶體和液體的中間狀態。然而,當時我沒有認識到這個新項目的價值。“什么是液晶?”知道了這個單詞的含義之后這個簡單的問題一直留在我的腦海里面。這是一個從零開始的研究(沒有任何相關的知識)。
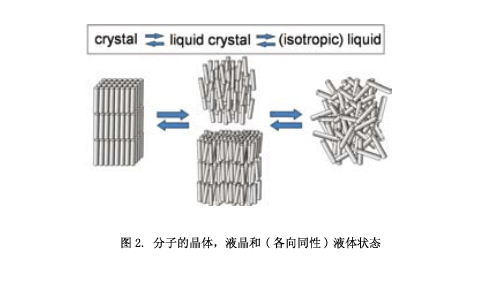
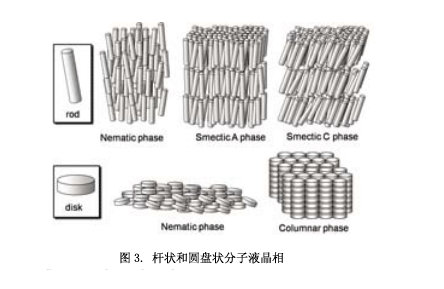
如圖2所示,液晶是晶體和液體(各向同性)之間的中間狀態。它兼有流動性和位置有序性的特點。桿狀和圓盤狀的分子具有液體的晶體。例如(圖3),類似棒狀分子的液晶相中,已知的有向列相、近晶A相、近晶C相;圓盤狀分子中液晶相有向列相和柱狀相。此外,還有三維光學各向同性的立方相。
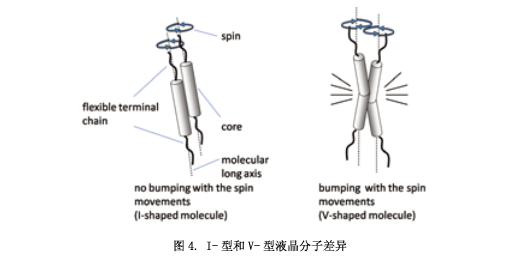
多數用于液晶顯示器的液晶分子具有一個像桿狀“I”的性狀結構(圖4左)。I狀分子圍繞著液晶相長軸快速旋轉,這個旋轉運動對液晶相的穩定有很重要的作用,通過高速的運轉阻止分子在低溫液晶范圍內結晶。然而,曲棒狀分子(V-型分子) (圖4右)不能夠順利的圍繞分子軸轉動,從而導致液晶相的不穩定性。
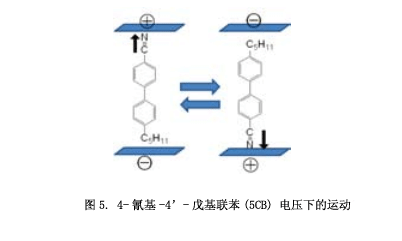
I型液晶分子在分子的末端具有一個極性取代基,這些分子會對電場作出回應。例如,4-氰基-4’-戊基聯苯(5CB) 用于液晶顯示器,施加電壓將會改變其場方向。如圖5所示 (箭頭表示分子偶極),通過電場,分子平行排列。分子長軸方向上的電極性不但能夠抑制分子的轉動,而且會形成一個定向的分子棒狀結構。然而,I型液晶分子具有一個橫向的分子極性,通過分子間的偶極-偶極相互作用抑制其旋轉,導致液晶相的不穩定性。特別是他的中心具有一個橫向的偶極分子,不能順利旋轉,很容易重結晶。
現在,我清楚了由Swager教授設計的這個分子對于液晶相的穩定性有兩處缺點(V型和中央橫向偶極子) 。這種分子構型對于制備穩定的液晶相是不合理的。這是一個非常具有挑戰性的項目,在麻省理工的前三個月,我合成了30種這種具有彎曲棒狀和中央偶極子的分子。這些分子理所當然的沒有顯示出任何的液晶相態,他們很容易就會結晶了。
與此同時,大量的實驗和失誤之后,我發現了一個暗示,能夠使具有V型和中央橫向偶極子的分子生成一個穩定的液晶相。錨和螺旋轉結構是產生穩定液晶相的必要條件1) 。曲桿和中心偶極像錨一樣停止分子轉動。但是如果分子中有螺旋結構轉動,那么它保持分子運轉抑制分子結晶,從而穩定液晶相1) 。螺旋結構與錨結構分離在穩定液晶相方面有很高的影響。
這個概念可以擴展延伸到其他具有強分子間相互作用的液晶分子的合成中。如果分子中的膜部分保持運轉、震動、滑動,那么這個分子就不會結晶從而保持液晶狀態。雖然這個概念不是很新穎的,我依舊在MIT 學習了一年的時間。在那之后, 我的項目就是選擇合成具有一個大的橫向影響作用的新型液晶分子的合成。這個概念基于“分子部分的持續移動”。
2. 分子中心橫向方向上偶極間的相互作用的介紹
化合物1和2在MIT合成的(圖6)2) 。這些分子中心部分是一個具有兩個氰基基團的噻唑環,并且有一個大的偶極矩(超過6 debye的計算值)。化合物1長度較短,液晶相不穩定,其中一個向列相在26℃,低于其熔點溫度時被觀測到會處于一種超級冷卻狀態。分子的相態從液晶態瞬間轉換為結晶態,因為他們具有彎曲的桿結構,短分子軸方向有一個很大的分子偶極子,分子旋轉受到強烈的阻礙。然而,令我驚奇的是化合物2,在1的兩個核心終端各引入一個苯環,表現出非常穩定的液晶相。它的向列相和近晶相A的相溫度范圍分別是160-252和135-160℃。這個液晶相的穩定性的改進主要是源于分子的終端引入了旋轉結構。縱橫比(=分子長度/分子的寬度)的增加,引入兩個苯環和兩個酯基增強了其分子間的相互作用,都被認為是其穩定性的原因。


化合物3(圖7a) 是我在完成MIT 項目后,在千葉大學合成的第一個液晶化合物3) 。它具有高度彎曲的形狀和一個大的偶極矩。這兩個棒狀介晶中心由-CO-NR-CO-(R=alkyl chain) 鏈接形成一個U型分子,具有12 debye的分子偶極。這個化合物在98-199℃顯示出穩定的近晶相A(圖7b).化合物3的X射線衍射圖中, 可觀測到兩個圖層的距離(圖7c)。一般情況下,近晶相A只有一層間隔。循環加熱和冷卻過后,這兩個隔層融合成為一個層距。這種分子具有很強的分子偶極子,分子間的相互偶極作用很強。這個層面臨近的分子不能夠在分子的長軸方向上順利的轉動,因為分子間強烈的相互作用。此外,如圖7d所示,每個分子有兩個介晶芯終端鏈,而化合物3在鏈接方向只有一個終端鏈。分子一端與另一端的烷基鏈空間平衡是不一樣的。因此可以認為是這種不平衡導致產生了兩種不同類型的層結構。一個相鄰層之間沒有有交錯的結構,一個相鄰層之間有交錯結構。兩層距離之間的差異與交叉烷基鏈的長度相對應。
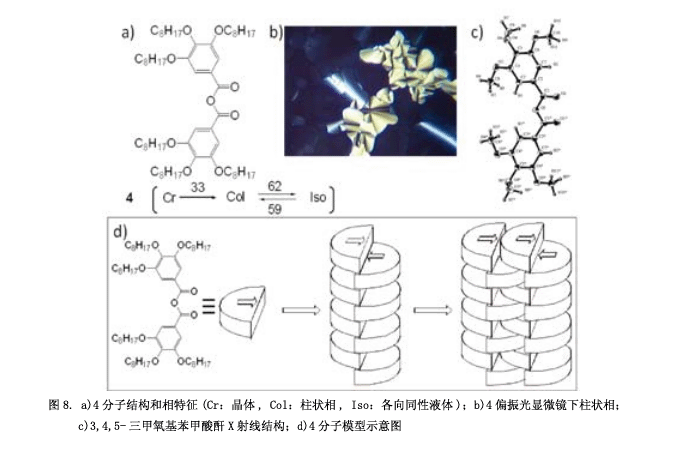
接著,一個更為簡單的分子(4)被設計合成,它具有大的偶極和彎曲的棒狀結構(圖8a)。它顯示一個柱狀的液晶相(圖8b) 4) 。在化合物4中的三個烷氧基能夠為苯環提供電子,羰基可以阻止水分子進攻羰基碳原子。因此,化合物4是一個穩定的酸酐,可以用柱層析法純化和重結晶。基于3,4,5-三甲基苯甲酸酐(圖8c)的X射線和其分子模型,分子間強大的偶極作用使得分子聚集成為一個柱狀分子團,成不平衡的并列狀構型(圖8d)。
3.橫向分子間氫鍵的介紹
氫鍵是分子間相互作用的強健。向列相和近晶相穩定能量約是1千卡/摩 (多數柱狀相是幾千卡/摩)。氫鍵的強度大約是1-9千卡/摩,其中大部分是2-3千卡/摩。多數情況下,氫鍵的終端有一個棒狀核心單位和氫鍵分子長軸方向平行5) ,在這種情況下,氫鍵不會抑制分子的旋轉和液晶的穩定。然而,在分子短軸方向上的分子間的作用抑制分子的轉動。
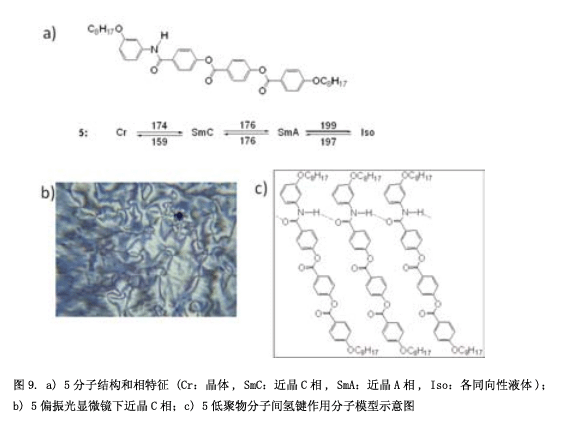
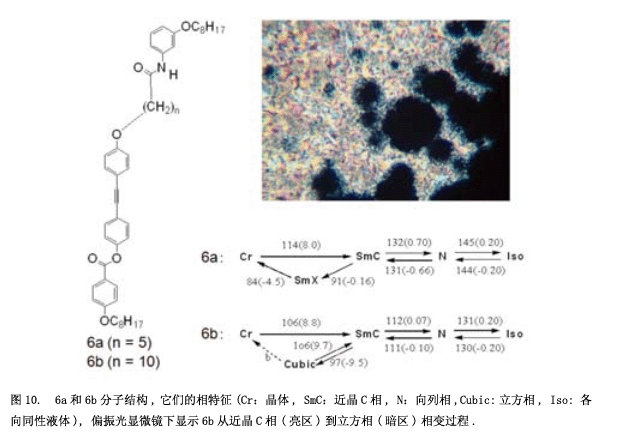
我們合成了一個具有橫向方向氫鍵的化合物5(圖9a),并研究其性質和分子堆積結構。在液晶相中,通過紅外光譜圖可以觀測到氫鍵6) 。液晶相的溫度范圍非常窄,特別是近晶相C更窄(圖9b)。假設是氫鍵阻礙了分子的轉動,液晶相變得不穩定(圖9c)。然后, 我們合成化合物6 (圖10)具有一個“錨結構”的強的氫鍵和一個“螺旋結構”持續轉動7) 。正與我們所料,化合物6顯示出穩定的向列相和近晶相C。此外,也可以觀測到一個立方相(圖10中照片)。令人驚訝的是,這個立方相顯示出自發的手性誘導作用,盡管這個分子并沒有手性。自發的手性誘導在晶相中經常能夠觀測到,但是一般情況下,這種現象在液體或者是液晶流體中出現。可以假設這種現象通過剛性分子間氫鍵構建起來。從我們之前的結果來看,也可以認為這種自發的手性還原源于酯基的扭曲構型8) 。盡管分子沒有任何手性,但是酯基(-CO-O-)在C-O單鍵和O鍵之間有一個左鍵或是右鍵,在第一階段, 具有左鍵和右鍵的分子生成1:1的比例,然后又同樣手性的分子組在一起成為一個分子聚集體。這種感應手性固定在每一個領域。此外, 我也計劃了將一個很強的氫鍵脲引入分子(圖11a)9) 。
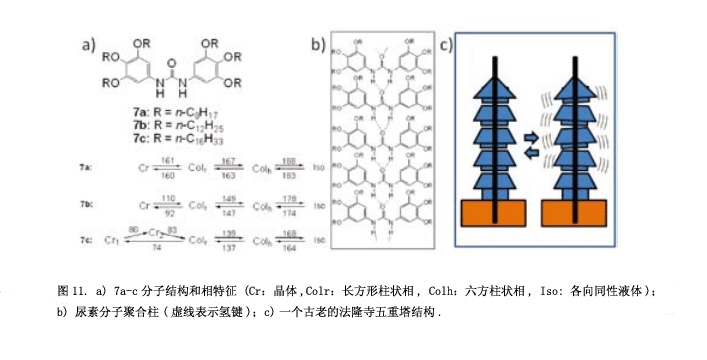
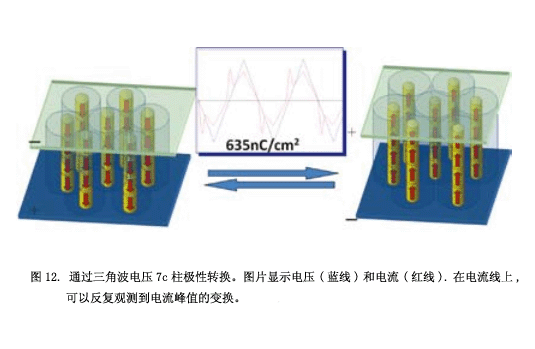
這個脲分子自行組成一個線性分子聚集體,如圖11b所示。化合物7a-c在它們分子末端有六個大的烷基鏈的,分子間的空間位阻斥力在相鄰的分子-NH-CO-NH-面之間產生了一個扭轉角,從而形成一個螺旋的柱狀結構。只有柱中心的部分有極性尿素分子,柱周邊的部分是沒有極性的烷基鏈。通常情況下,相鄰的極性柱有一個反-平行的排列可以取消她們的極性和高穩定性而不改變液晶相的極性方向。然而, 在7a-c的結構中,柱狀間的偶極相互作用遠遠小于其他液晶化合物柱,因為偶極間柱結構間的的長距。另外,中央線性尿素分子的重復距離是4.7?,它們的苯環因為太長距離4.7?,而沒有強的分子間相互作用。這種結構類似于一個古老的法隆寺五重塔(圖11c),這是世界上最古老的木結構建筑。所有的樓層都由貫穿中心寶塔的一個長而大的支柱支撐。當地震發生時,這座塔不會倒塌因為中心柔軟的木質支柱。我們的尿素分子中心柱也有一個強大的線性氫鍵網結構,而分子的其他部分在相鄰的分之間沒有強的分子間作用,因此這個中心柱結構柔軟并且晃動。因此,這寫極性分子在靈活的極性柱配合下將他們的偶極子從一個方向轉變到另一個方向,因此柱結構的極性可以改變方向(圖12)。這個圖片顯示了當前的變化,并且應用一個三角波電壓和合成7c液晶相中的細胞電容器。每一個分子的定向變化源于尿素分子的構象變化。目前為止,有幾篇關于柱狀相手性分子極性切換的報告10),但是沒有柱狀相非手性分子極性切換的例子11) 。我們關于這個轉換性能的報道后,一些科學家聲稱轉換峰源于離子雜質的運動。后來,柱極性轉換通過二次諧波的產生實驗證明12) 。
現在,我們正試圖通過增加柱狀間的距離或是增加轉換能源缺口使得極性狀態穩定。在不久的將來,我們將會按列控制柱的極性方向來實現高密度的存儲裝置。
4. 橫向方向上全氟芳烴-芳烴間相互作用的介紹
1960年, Patrick和Prossor發現了六氟苯(mp 5.0 ℃) 和苯室溫下(mp 5.4 ℃) 按1:1比例配合得到結晶 (mp 23.7 ℃),這個發現之后開創了全氟芳烴-芳烴間相互作用的歷史13)。這個影響由范德華力和靜電相互作用而成,能量是3.7-5.6千卡/摩14),這幾乎與其中的氫鍵相同水平。據我們所知,通過全氟芳烴-芳烴間相互作用得到穩定的液晶相,只有兩個例子是眾所周知的15) 。在我們的實驗室里,簡單的化合物8具有一個全氟苯基和一個三烷氧基苯基(圖13a) 16) 。
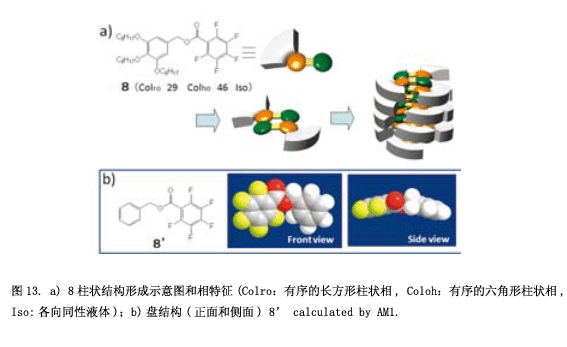
 如我們所料,這些分子在不同的π-面產生強烈的全氟芳烴-芳烴間相互作用,表現出柱狀相。因為真空中分子8’(圖13b)彎曲的桿狀結構,產生了一個我們預計的宏觀意義上可見的極性柱。然而,運用三角波電壓下我們并沒有觀測到極性現象,這可能是因為分子間強的全氟芳烴-芳烴間相互作用的抑制。也有可能,每一個分子的柱結構與其相鄰的分子中心部分(如圖7所示的柱狀結構)有極性轉換的必要。在聚甲醛中,顯示出很強的分子間作用,并且可以觀測到磁帶裝紋理(圖14a)。從二維XRD (圖14b)可以看到,堆積在柱周邊的分子組成了磁盤。
如我們所料,這些分子在不同的π-面產生強烈的全氟芳烴-芳烴間相互作用,表現出柱狀相。因為真空中分子8’(圖13b)彎曲的桿狀結構,產生了一個我們預計的宏觀意義上可見的極性柱。然而,運用三角波電壓下我們并沒有觀測到極性現象,這可能是因為分子間強的全氟芳烴-芳烴間相互作用的抑制。也有可能,每一個分子的柱結構與其相鄰的分子中心部分(如圖7所示的柱狀結構)有極性轉換的必要。在聚甲醛中,顯示出很強的分子間作用,并且可以觀測到磁帶裝紋理(圖14a)。從二維XRD (圖14b)可以看到,堆積在柱周邊的分子組成了磁盤。
5. 聚合近晶相分子模型的應用
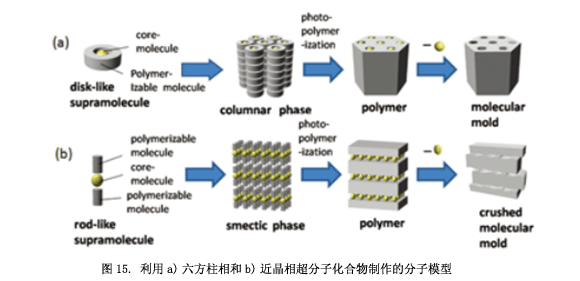
如圖15a所示, 這是一個用于制造分子模具非常有用的六方柱狀相17)。柱狀構型的聚合在它們中心部分具有核心分子(或者離子),然后去除中心分子(或者離子)得到納米多孔材料。這種材料有一個剛性的蜂窩狀結構,對分子識別非常有用。另一方面,利用近晶相制作分子模型非常困難(圖15b),從相應的聚合物中去除核心分子會導致層結構受到擠壓。
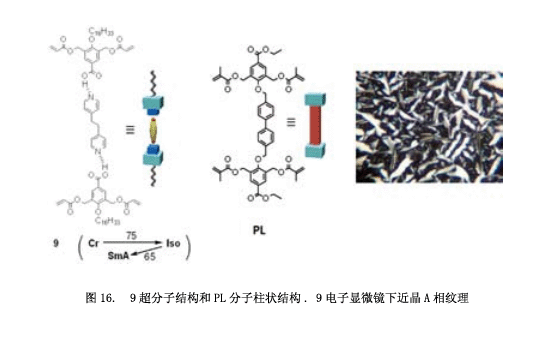
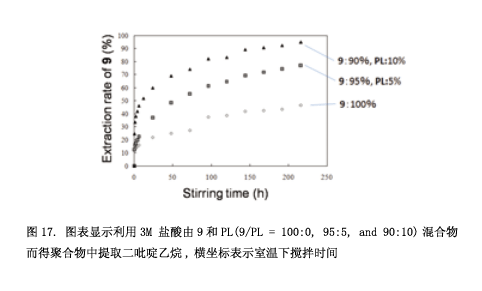
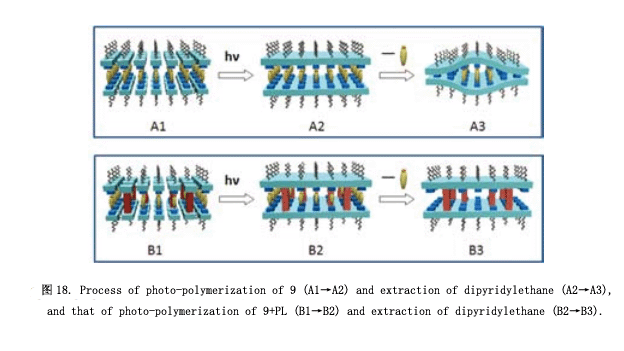
盡管如此,我們仍試圖用近晶相合成分子模型18) 。如圖16所示,超分子9 有一個二吡啶乙烷基分子作為核心分子,每一個終端通過氫鍵連接著一個p-烷氧基苯酸衍生物。兩個聚合取代基橫向引入到這個本算衍生物里面。我們也合成了一個桿狀化合物PL ( “pillar”的縮寫) ,其中的分子部分共價連接。化合物9展現出近晶相A (如圖16微照片) ,這些分子里面有一個多層次的超晶格結構,純化合物(9: 100%)光聚合得到相應的聚合物。然而,如圖17所示,利用3M的鹽酸從該聚合物中提取二吡啶乙烷非常緩慢。即使是216小時之后,仍有50%的二吡啶乙烷留在聚合物中。假設因為聚合物邊緣層結構受到擠壓使得這個核心分子不能夠出去(圖18: A1→A2→A3).然后,我們嘗試著將PL分子引入分子層。將5% 的化合物9和10% 化合物PL混合得到相應的混合物,混合物光聚合為液晶相。從這個聚合物中提取核心分子,可以分別獲得75% 和100% 核心分子。因此,這證實了近晶相超晶格結構通過引入PL可以用于制作分子模型。我們將要合成一種聚合物,通過引入PL分子與客體分子連接一起使得它具有一種很高的分子識別能力。
6.總結
從我得到一張寫有關于液晶分子的文章已經12年過去了。我一直都在研究具有強烈橫向互動的液晶分子。在分子的短軸方向上引入一個強大的作用力可以強烈的破壞液晶態的穩定,并且在多數情況下不會顯示任何的液晶相。然而,如果我解決這些問題,我可以有很多機會來找到新的現象。并且我也可以極大地感覺到一種成就感。這些年,我有一個策略就是“在我的項目中,用最簡單的分子可能來完成目的”,優點之一是,簡單的化合物很容易得到,更為顯著的優點是以簡單分子合成目的會給許多科學家以強烈的沖擊,并且他們中會有很多項目研究用到我們的分子。更進一步說,我們可以很容易的用簡單的分子來解釋一個新穎的現象,并且可以很容易的想象出它的應用。所以,我一直致力于研究變異的和簡單的液晶分子的合成并從中找到有用的分子。
相關文獻
1) K. Kishikawa, N. Muramatsu, S. Kohmoto, K. Yamaguchi, M. Yamamoto, Chem. Mater. 2003, 15, 3443.
2) K. Kishikawa, M. C. Harris, T. M. Swager, Chem. Mater.1999, 11, 867; S. H. Eichhorn, A. Paraskos, K. Kishikawa, T. M. Swager, J. Am. Chem. Soc. 2002, 124, 12742; I. A. Levitsky, K. Kishikawa, S. H. Eichhorn, T. M. Swager, J. Am. Chem. Soc. 2000, 122, 2474.
3) K. Kishikawa, Y. Miwa, T. Kurosaki, S. Kohmoto, M.Yamamoto, K. Yamaguchi, Chem. Mater. 2001, 13, 2468; T. Kajitani, Y. Miwa, N. Igawa, M. Katoh, S. Kohmoto, M. Yamamoto, K. Yamaguchi, K. Kishikawa, J. Mater. Chem.2004, 14, 2612.
4) K. Kishikawa, S. Furusawa, T. Yamaki, S. Kohmoto, M.Yamamoto, K. Yamaguchi, J. Am. Chem. Soc. 2002, 124,1597.
5) Reviews: T. Kato, N. Mizoshita, K. Kishimoto, Angew.Chem. Int. Ed. 2006, 45, 38; T. Kato, T. Yasuda, Y. Kamikawa, M. Yoshio, Chem. Commun. 2009, 729-739.
6) T. Kajitani, S. Kohmoto, M. Yamamoto, K. Kishikawa, J.Mater. Chem. 2004, 14, 3449; T. Kajitani, S. Kohmoto, M. Yamamoto, K. Kishikawa, Chem. Mater. 2004, 16, 2329; T. Kajitani, S. Kohmoto, M. Yamamoto, K. Kishikawa, Mol. Cryst. Liq. Cryst. 2005, 439, 2039.
7) T. Kajitani, S. Kohmoto, M. Yamamoto, K. Kishikawa,Chem. Mater. 2005, 17, 3812.
8) T. Kajitani, H. Masu, S. Kohmoto, M. Yamamoto, K.Yamaguchi, K. Kishikawa, J. Am. Chem. Soc. 2005, 127,1124; S.-W. Choi, K. Fukuda, S. Nakahara, K. Kishikawa, Y. Takanishi, K. Ishikawa, J. Watanabe, H.Takezoe, Chem. Lett. 2006, 35, 896; S. Kawauchi, S.-W. Choi, K. Fukuda, K. Kishikawa, J. Watanabe, H. Takezoe, Chem. Lett. 2007, 36,750.
9) K. Kishikawa, S. Nakahara, Y. Nishikawa, S. Kohmoto, M. Yamamoto, J. Am. Chem. Soc. 2005, 127, 2565; H. Takezoe, K. Kishikawa, E. Gorecka, J. Mater. Chem. 2006,16, 2412.
10) H. Bock, W. Helfrich, Liq. Cryst. 1992, 12, 697; H. Bock, W. Helfrich, Liq. Cryst. 1995, 18, 387; H. Bock, W. Helfrich, Liq. Cryst. 1995, 18, 707; G. Heppke, D. Krüerke, M. Müller, H. Bock, Ferroelectrics 1996, 179, 203; G. Scherowsky, X. H. Chen, Liq. Cryst. 1994, 17, 803; G. Scherowsky, X. H. Chen, J. Mater. Chem. 1995, 5, 417; J. Barbera, R. Iglesias, J. L. Serrano, T. Sierra, M. R. de la Fuente, B. Palachios, M. A. Perez-Jubindo, J. T. Vazquez, J. Am. Chem. Soc. 1998, 120, 2908.
11) L. Lei, Mol. Cryst. Liq. Cryst. 1983, 91, 77; L. Lei, Mol. Cryst. Liq. Cryst. 1987, 146, 41; A. M. Levelut, J. Malthete, A. Collect. J. Phys. (France) 1986, 47, 351; H. Zimmerman, R. Poupko, Z. Luz, J. Billard, Z. Naturforsch., Teil A 1985, 1794; B. Xu, P. J. Carroll, T. M. Swager, Angew. Chem. Int. Ed. 1996, 35, 2094; M. Sawamura, K. Kawai, Y. Matsuo, K. Kanie, T. Kato, E. Nakamura, Nature
2002, 419, 702.
12) Y. Okada, S. Matsumoto, Y. Takanishi, K. Ishikawa, S.Nakahara, K. Kishikawa, H. Takezoe, Phys. Rev. E 2005,72, 020701(R); Y. Okada, S. Matsumoto, F. Araoka, M. Goto, Y. Takanishi, K. Ishikawa, S. Nakahara, K. Kishikawa, H. Takezoe, Phys. Rev. E 2007, 76, 041701.
13) C. R. Patrick, G. S. Prosser, Nature 1960, 187, 1021.
14) O. R. Lozman, R. J. Bushby, J. G. Vinter, J. Chem. Soc.Perkin Trans. 2 2001, 1446; S. Lorenzo, G. R. Lewis, I. Dance, New J. Chem. 2000, 24, 295; J. HernIndez-Trujillo, F. Colmenares, G. Cuevas, M. Costas, Chem. Phys. Lett. 1997, 265, 503; A. P. West, Jr., S. Mecozzi, D. A. Dougherty, J. Phys. Org. Chem. 1997, 10, 347.
15) M. Weck, A. L. Dunn, K. Matsumoto, G. W. Coates, E. B.Lobkovsky, R. H. Grubbs, Angew. Chem. Int. Ed. 1999,38, 2741; C. Dai, P. Nguyen, T. B. Marder, A. J. Scott, W.Clegg, C. Viney, Chem. Commun. 1999, 2493.
16) K. Kishikawa, K. Oda, S. Aikyo, S. Kohmoto, Angew.Chem. Int. Ed. 2007, 46, 764.
17) H.-K. Lee, H. Lee, Y. H. Ko, Y. J. Chang, N.-K. Oh, W.-C.Zin, K. Kim, Angew. Chem. Int. Ed. 2001, 40, 2669.
18) K. Kishikawa, A. Hirai, S. Kohmoto, Chem. Mater. 2008,20, 1931.
注:本文為提供者翻譯的,由于知識所限,其中錯誤在所難免,敬請原諒。如有問題可以查找原文。
快速導航
化學品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
關于物競
物競數據庫是一個全面、專業、專注,并且免費的中文化學品信息庫,為學生、學者、化學品研究機構、檢測機構、化學品工作者提供專業的化學品平臺進行交流。
數據庫采用全中文化服務,完全突破了中英文在化學物質命名、化學品俗名、學名等方面的差異,所提供的數據全部中文化,更方便國內從事化學、化工、材料、生物、環境等化學相關行業的工作人員查詢使用。
關注我們

-
微信賬號:物競化學品數據庫

-
微博賬號:wjhxp
聯系我們
上海市延長路149號上海大學科技園412室
公司總機: 021-56389801
訂購電話: 4007001514
傳真電話: 021-56389802
客服電話: 021-56332350
電子郵件: wingch@basechem.org
 滬公網安備 31010602001115號
滬公網安備 31010602001115號