氧合銨鹽氧化醇
- 2012-12-31
- 專題
1.介紹
這篇文章中的多數材料已經在最近一篇題為“氧合銨和氮氧催化氧化醇”的專題中進行了一個全面的討論。1)兩篇先前的關于氧合銨2)和這個項目中所用的實驗方法3)也被使用過。這篇報道中所述的氧化反應(多數是醇的氧化)含有一個明顯的氧合銨陽離子選擇性。即便是有,極少數情況下可以觀測到異構化現象,無論是雙鍵(順式-反式)還是手性中心,也不會發生碳碳鍵裂解。此外,這些試劑都是“綠色環保”的沒有重金屬鉻或是錳的參與。
Eq.1顯示該反應過程。
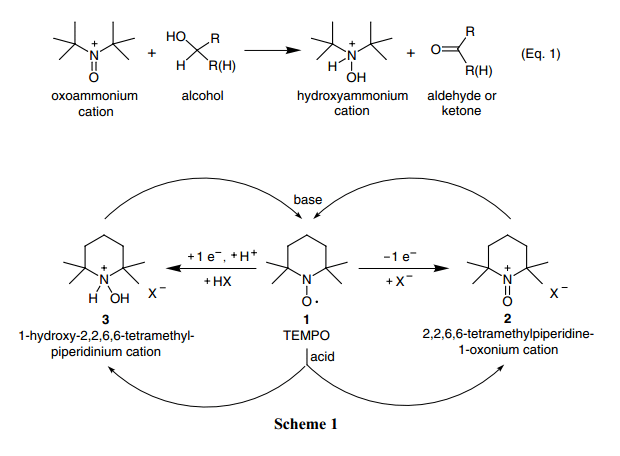
對于氧銨離子是穩定的,可以沒有碳氫原子(α-氫原子)連接氮, 也滅有必要因為其他的原因在氮和相鄰碳原子間形成一個雙鍵。4)這里有很多這類鹽的例子,2)但是常見的多數都是基于哌啶鹽基礎上的,如Scheme1所示。在Scheme1里面,氮氧1, 氧銨鹽2, 羥銨或是其鹽3的氧化還原性能和其制備、分解的常規方法被總體概括。
這個項目中的每一部分最知名的化合物就是TEMPO或者2,2,6,6-四甲基哌啶氧化物1。TEMPO是一個穩定的氧自由基,發現于1962。5)合適的陰離子的參與下,從TEMPO中移除一個電子生成氧銨鹽,2 (2,2,6,6-四甲基哌啶-1-氧鎓或者2,2,6,6-四甲基-1-氧代哌啶鎓鹽)。合適的陰離子和算條件下,TEMPO加入電子得到羥銨鹽3 (1-羥基-2,2,6,6-四甲基哌啶鎓鹽)。在強酸中發生顯著的歧化反應,TEMPO被轉化為2和3中的一個分子形式。這個反應由Golubev發現,并且作為制備氧銨鹽的方法之一。6)該反應強大的驅動力幾乎可以肯定來自于中性TEMPO離子產物的形成。如果2和3混合物被制成堿性,那么就會有相對應的反歧化反應發生,從而將化合物重新轉換為1。
結合Eq.1和Scheme1, 很明顯,醇氧化涉及了氧銨鹽2中一個二電子反應,轉換為羥銨鹽3。氧銨離子氧化反應可以用四個途徑(Scheme 2);在化學計量氧化反應中,酸(Eq.2)或是堿(Eq.3), 再或者利用酸-歧化反應(Eq. 4)如Scheme1所述,和利用輔助氧化劑比如次氯酸鈉(漂白劑)氮氧催化氧化(Eq.5)。這些方法中,使用氮氧和酸的酸歧化反應(Eq.4)少有研究。7,8)氮氧催化反應(Eq.5)研究廣泛,并可以用到很多的不同的二級氧化劑,1) 但是這篇文章中也不做考慮。式2-5中,R基團可以是氫或者其他的基團。
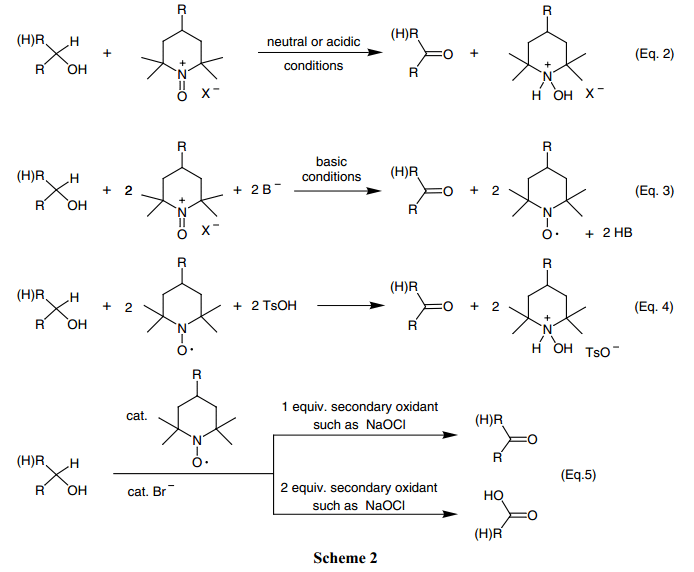
我們主要對化學計量反應感興趣,即預備氧銨鹽反應中使用1mole/1mole當量的醇或者其他化合物被氧化。這些反應可以在堿的參與下于中性或者是微酸性條件下進行,如Eqs. 2和3所示。Eq.4中所描述的酸歧化反應是一個化學計量法過程,在這個反應中氮氧化物和p–苯甲磺酸(兩個當量)用于化學計量法中的量。
2.氧銨鹽的制備及其性能研究
通過進一步氧化氮氧化物例如1來制備氧銨鹽。反過來,氮氧化物的制備是通過氧化不含有α–氫或者是不能形成雙鍵的胺來完成。很多已知的氮氧化物具有不同的結構,9-11)因此有很多種類的氧銨鹽。2)然而,幾乎所有已報道的氧化反應都是通過由2,2,6,6–四甲基哌啶衍生而得的氮氧化物(作為催化劑或者鹽)來進行完成的。這主要歸因于它們的商業可用性。最常用的鹽如Scheme 3所示。相對應的氮氧化物被用于酸岐化或者是氮氧催化反應中。
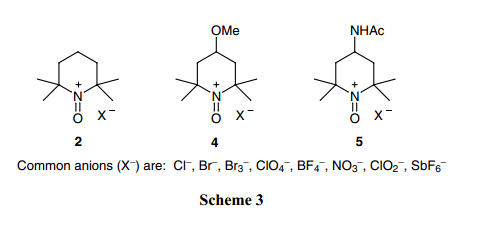

市面上唯一的氧銨鹽(5作為四氟硼酸鹽)的總合成如Scheme 4所示。Bobbitt12)文章中給出了5(BF4?)的具體說明和其有機合成的描述。13)
Scheme 4中的反應根本上是在Scheme 1 Golubev6)中工作基礎上得來的。合成氧銨鹽其他的主要方法涉及到用鹵元素氧化氮氧化物從而得到鹵代鹽。14)
化合物6(4-乙酰氨基-2,2,6,6-四甲基哌啶-1-氧化物)需要特殊的標注,尤其是與TEMPO進行比較時。它的熔點是143-145°C,相對于36-38°C的TEMPO,更加穩定。它很容易從4-氨基-2,2,6,6-四甲基哌啶(Scheme 4)中制備,并且代替TEMPO廣泛的應用在氮氧化物催化氧化反應中。它還具有十分有趣的溶解特征,極易溶于二氯甲烷,部分溶于水,微溶于乙醚。因此,它可以用二氯甲烷將其從水中提取出來,或是用水將其從乙醚中提取出來。可以從乙酸乙酯或水中重結晶(損失很少)。
3.醇在酸性或是近中性條件下的氧化
在由Bobbitt12)所報道前于所發現的高氯酸鹽的一系列的醇類和5(ClO4?)的氧化反應是不穩定的,很容易爆炸。15)然而在同樣的一篇報道中市售四氟硼酸鹽,似乎功效非常好。Scheme 5中顯示了用硅膠作為催化劑催化氧化醇的整體反應方案。

醇漿,明黃色的氧銨鹽和硅膠在二氯甲烷中攪拌直至顏色變白。將混合物過濾,通過硅膠薄墊,然后蒸發至干。通常,沒有必要進一步的純化。產率很高,并且二氯甲烷非常適合一些后續的反應 (格氏反應, Wittig反應, Baylis-Hillman反應和其他更多中反應)而不需要產品隔離。該方法用于制備低分子量或者是不穩定的醛、酮十分理想。
這個反應具有很多優點。他在室溫下二氯甲烷中進行,并且不要求嚴格的無水條件。就意義上而言,這個反應是比色反應,混合物從明黃色漿液變至白色漿液。這個鹽充分可溶,并且在溶劑中反應,但是氧化還原劑7 , 極不易溶。硅膠和7混合物處理過后得到回收物6。12,13)
然而正常的從醇到羰基化合物的氧化反應,二醇有時候會得到內酯,尤其是在五元六元環形成的時候。16,17)在吡啶(堿反應)的參與下也可以生成內酯,有一些具有大環結構。18,19)
各種醇類氧化反應速率是,從最快到最慢依次是芐基或烯丙基醇,仲脂肪醇和炔醇,最后是伯脂肪醇。12)在硅膠的參與下,所有的反應速率都顯著加快。
硅膠催化的原因尚不明確,但是可以通過想象來解釋,極性硅膠吸收極性分子在其表面上,因此相對增加了反應物的局部濃度。有一個類似的催化劑被指出是氧化鋁,而不是極性小的木炭。12)從實際應用的角度出發,50-50的鹽和硅膠混合反應就可以進行。將混合物或鹽研磨,如果單獨使用,反應速率似乎會加快。這可能是因為這個反應是一個表面反應或者僅僅是細碎的試劑更快的溶于溶液。硅膠吸附少量的副產物有助于產品的隔離。
這個方法的主要缺點是具有β氧功能的醇的氧化很慢使得反應不進行。7,12)盡管這個抑制作用的原因尚不明確,但是這樣的分子內部氫鍵可以產生一個五元環,這就可能會抑制該反應反應機制中可視化氫鍵中間體的形成(參見酸性或中性介質反應機制)。
除了少數情況例外(參閱非醇類-氧化反應), 胺與氧銨鹽反應得到無法識別的產品并且很好避免,盡管胺性質穩定。分子中具有一個三取代雙鍵、芐氧基或者縮醛基團時與試劑反應十分緩慢。保護集團叔頂級二甲基硅烷氧基(TBDMS)緩慢裂解,但是叔丁基二苯基硅烷氧基(TBDPS)穩定。12)三取代基烯烴與乙腈的反應有了進一步的研究,20)同樣的與芐基的反應也是。21,22)唯有與脂肪醇的反應是有問題的;芐基或烯丙基醇在二氯甲烷中反應迅速并且沒有任何問題。通常在堿性介質中發生的氧化催化反應很顯然不會存在這些缺點。
4.基礎條件下氧銨鹽的氧化
氧銨鹽堿性條件下在水中反應得到過氧化物和氮氧化物。23)重要的是要注意,幾乎所有的氮氧化物催化氧化反應都發生在水環境下的堿性介質中。1)
在吡啶堿的參與下,反應在二氯甲烷中反應也會發生,然而這個反應少有研究。18,19,24)反之,具有β氧的醇類與氧銨鹽在中性或酸性條件下不反應,在吡啶的參與下它們能夠得到收益很好的二聚體酯。18) Eq. 6中顯示了糖衍生物一個具體的例子。反應允許烯丙基雙鍵、丙烯酸酯、芐氧基、環丙基醚和硫化物的存在。
該反應似乎取決于吡啶堿的性質,因為2,6-lutidine參與下形成了醛。25)這些反應正在研究中。
5.通過酸岐化方法的氧化
酸岐化反應中,如Eq.4所示,一當量的基質,兩當量的氮氧化物6,和兩個當量的p–甲苯磺酸加入二氯甲烷中攪拌。7)氮氧化物歧化得到一個當量的氧銨甲苯磺酸酯和一個當量的羥胺甲苯磺酸酯,如Scheme 1所示。氧銨鹽甲苯磺酸酯氧化反應轉換為一個兩個當量的羥銨甲苯磺酸酯析出。混合物過濾,濾液用水和鹽水洗滌,干燥,必要時進一步純化。像氧銨鹽氧化反應屬于比色法,橙色的氮氧化物被轉換為白色漿液,產率很高。7)此外,在氧銨鹽反應中,濾去的氧化劑可以再生得到氮氧化物。

在澳大利亞,這個方法已經被Banwell 團隊廣泛的開發研究。8,26)在Banwell的研究中,1,2-二醇很容易氧化成羥基酮或者1,2-二酮(Scheme 8)。很顯然,這些反應中沒有β氧抑制劑。
6. 機制
醇氧化為醛或酮是一個看似簡單額轉換反應。關于醇類的氧銨鹽氧化反應和氮氧催化氧化反應機制已經有大量的研究公布出來,雖然兩個質子和兩個電子(Eq. 1)的方式的資料還沒有完全的清楚。27-29)
在本節中,我們將只涉及包括氧銨陽離子和各種基質材料的機制么。催化機制竟在其他地方描述。1)
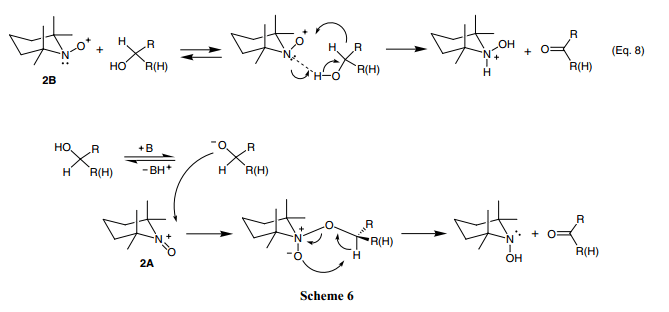
機制的某些不確定性源于氧銨陽離子可以配制出兩個反應共振形式,2A和2B (Eq.7)。2A 中有一個氧和氮完整的電子對,氮原子上面具有正電荷作為電負性原子。因此,這有助于實現其反應性能。然而,2B形式的存在也不能排除在外,代表了一個氧電子的少見的例子。30,31)2B可以用來解釋氧銨鹽與格氏試劑32)的反應和激活雙鍵(將討論在Miscellaneous副反應中不良反應),7,20,31,33),更重要的是很可能是醇氧化作用比迄今為止更容易實現。
酸或中性介質中的機制 在最初的脂肪醇計量化學氧銨鹽氧化反應中,基質的相對反應性能依次是仲醇>伯醇>甲醇,27,34)并且相應的反應速率與羥基軸碳上面已知的碳氫鍵的產度相吻合。抽象的氫化物被提了出來。
這種類型的機制,其中通過B3LYP/6-31+G*的能量計算,與異丙醇的氧化速率比甲醇的快是保持一致的。29)
環狀中間體,很可能借助2B結構中醇和氮電子對中氫之間的氫鍵的形成來促進該反應(Eq. 8).
氧銨鹽的反向陰離子對氧化反應速率有影響。14,35)氯離子的出現導致反應速率與高氯酸和四氟硼酸鹽離子相比反應速率更快,其中相應的,比溴離子速率也相對加快。Eq. 8中間體 忽略了任何陰離子的影響并且無法解釋所觀測到的反應速率的差異。在硅膠的存在下,使用高氯酸或者四氟硼酸鹽使得反應速率明顯提高。12)
酸性介質中,含有一個β氧的醇對于氧銨鹽的反應性的缺失,可能涉及到上面所提到的氫鍵。7,12,36)如果被氧化的羥基集團之間的氫鍵和氧化劑通過β氧化合物中的β氧和分子內氫鍵建合作用而被抑制,反應將會被制止或者顯著放慢速度。另一方面,這樣一個機制是否也將貫穿在強酸或者水性介質中是有疑問的。這個問題主要在堿性介質中進行的話似乎并沒有發生催化反應。
獲得的結果與氧銨鹽進行酸岐化反應增加了反應的混亂度。氧化反應中使用氮氧化物與p–甲苯磺酸作為歧化試劑(Eq. 4),β氧抑制簡單的分子,7)雖然他不一定適合更為復雜的案例。8)這一切表明,β氧效果還是一個謎。
堿性介質中的機制 幾乎所有的氮氧化物催化氧化反應都在堿性介質中進行。氧銨鹽與稀堿水溶液反應緩慢生成氮氧化物和過氧化氫。37,38)

在堿中的氧化反應被認為是含有作為一個親和物種的醇化物。27-29)醇的形成和反應進一步細節如Scheme 6所示。這種機制已經通過計算結果得到支撐。29)
這些化合物形成的平衡常數減少了醇鹽空間體積的增加,并且合成體的穩定性差異可以用來解釋所觀測到的伯醇比仲醇氧化速度快的現象。這是試驗中觀測到27),在氮氧催化反應中尤其重要。1)吡啶堿的存在下氧化反應化學計量研究已經被報道,但是機制尚不明確。18,24)
7.氧化反應的具體實例
很多氧銨鹽氧化反應(至少1,500種),包括化學計量和催化都已經有報道。1,2,39,40)在我們的有機合成反應章節中至08年7月,已經編目了270個化學計量氧化的例子(Eqs.2-4)。1)由于高氯酸鹽易爆,15)我們建議使用四氟硼酸鹽。
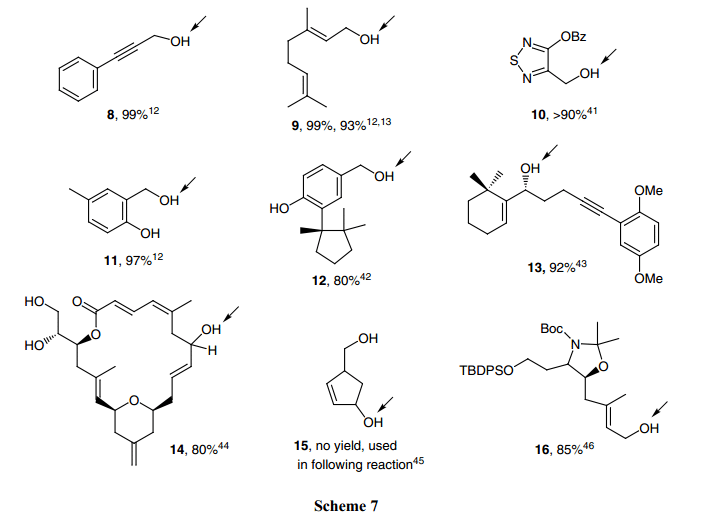
氧銨鹽氧化反應(Eqs. 2和3) Scheme 7中給出了一些更為復雜的基質的結構。氧化位置如箭頭所示,氧化得到羰基,可以是醛也可以是酮。反應顯示了在氧銨鹽氧化反應中可觀測到的某些選擇性。
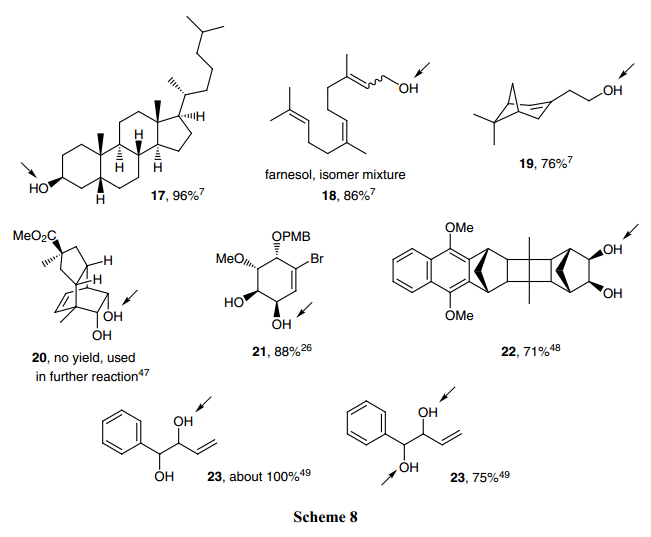
氮氧化物酸歧化氧化反應(Eq.4) Scheme 8中給出了幾個不均衡型氧化反應的例子。所用的酸是p-甲苯磺酸,反應在二氯甲烷中進行。
8.非醇類氧化化學計量
這里有幾個不是醇類氧化的氧銨鹽反應。在某種情況下,它們和醇類氧化可能會產生問題,但是多數發生在其他溶劑中,比如乙腈,在二氯甲烷中反應緩慢。
胺類與氧銨鹽的反應最好避免,且沒有實際用處。34,50,51)還有更多的工作需要去完成。
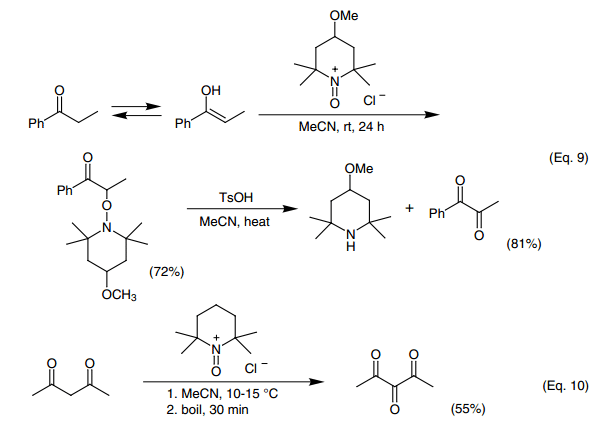
單酮可以被氧化為α-二酮(Eq.9),33,52-55)1,3-二酮可以被氧化成1,2,3-三酮(Eq.10)。53)后者反應生成1,2,3-三羰基化合物,不需要碳碳鍵的裂解,反應獨特,并沒有被進一步開發研究。
烯醇醚和烯胺與氧銨鹽反應得到加成產物。56,57)
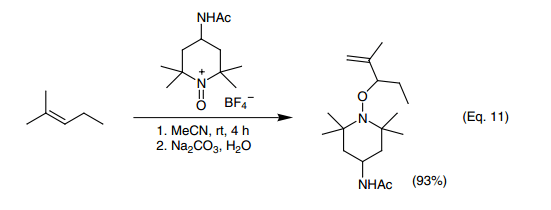
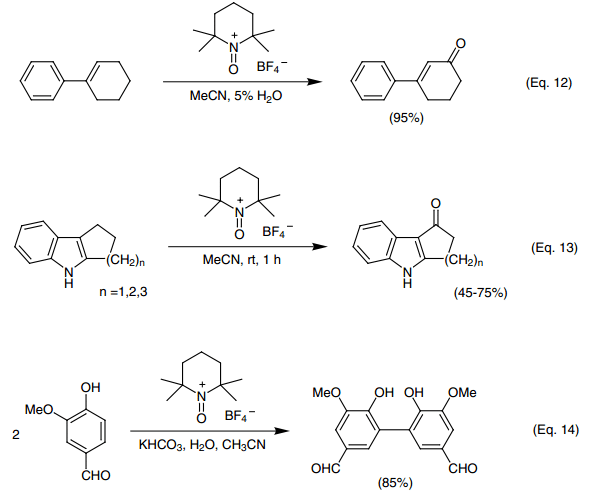
三烷基和四烷基二錫氧烷與氧銨鹽在二氯甲烷中反應緩慢,但是在乙腈中反應迅速并且得到加成產物(Eq. 11)。20)在一個獨特的反應中,一個活化的雙鍵生成不飽和酮(Eq. 12)。58)這個反應將得到進一步的探討。
在另一個獨特的反應中,氧銨鹽氧化一個碳附著于吲哚3位置處(像四氫咔唑和其類似物)得到酮(Eq. 13)。59)這個反應包含一個水分子的加成,但是不確定。
酚與苯基醚與氧銨鹽反應得到醌或者碳碳耦合二聚體(Eq.14)。51,60-63)
芐基醚在乙腈中反應迅速,得到苯甲醛,進一步氧化得到醇(Eq.15)。21,22)該反應在二氯甲烷中反應非常緩慢。在二氯甲烷中芐基醇的氧化速度非常的快使得芐醚(如果存在)不會受到影響。
硫化物與氧銨鹽的反應是有爭議的。硫醇與氧銨鹽反應得到二硫化物。56)據報道硫化物是具有抗氧化性的。60)二甲亞砜與氧銨鹽反應快速(比如在NMR tube中), 但是反應產物沒有明確。64)
9. 結語
氧銨鹽氧化和酸岐化氧化表現出高產率醇的簡單氧化和產品的隔離。此外,這個反應不要嚴格的反應條件,綠色且沒有重金屬。當產物有揮發性和不穩定時,這個反應尤其的合適。
References
1 J. M. Bobbitt, C. Brückner, N. Merbouh, Org. React. 2009, 74, 103-424.
2 J. M. Bobbitt, M. C. L. Flores, Heterocycles 1988, 27 , 509-533.
3 N. Merbouh, J. M. Bobbitt, C. Brückner, Org. Prep. Proc. Int. 2004, 36 , 3-31.
4 M. Shibuya, M. Tomizawa, I. Suzuki, Y. Iwabuchi, J. Am. Chem. Soc. 2006, 128, 8412-8413.
5 M. B. Neiman, é. G. Rozantzev, Y. G. Mamedova, Nature 1962, 196, 472.
6 V. A. Golubev, R. I. Zhdanov, V. M. Gida, E. G. Rozantsev, Russ. Chem. Bull. 1971, 20, 768-770. (English translation)
7 Z. Ma, J. M. Bobbitt, J. Org. Chem. 1991, 56 , 6110-6114.
8 M. G. Banwell, V. S. Bridges, J. R. Dupuche, S. L. Richards, J. M. Walter, J. Org. Chem. 1994 , 59 , 6338-6343.
9 é. G. Rozantsev, V. D. Sholle, Synthesis 1971, 401-414.
10 é. G. Rozantsev, V. D. Sholle, Synthesis 1971, 190-202.
11 J. F. W. Keana, Chem. Rev. 1978, 78 , 37-64.
12 J. M. Bobbitt, J. Org. Chem. 1998, 63 , 9367-9374.
13 J. M. Bobbitt, N. Merbouh, Org. Synth. 2005, 82 , 80-86.
14 T. Miyazawa, T. Endo, S. Shiihashi, M. Okawara, J. Org. Chem. 1985, 50 , 1332-1334.
15 J. M. Bobbitt, Chem. Eng. News 1999, 77 , 6.
16 T. Miyazawa,T. Endo, J. Am. Chem. Soc. 1985, 50 , 3930-3931.
17 P. L. Anelli, S. Banfi, F. Montanari, S. Quici, J. Org. Chem. 1989, 54 , 2970-2972.
18 N. Merbouh, J. M. Bobbitt, C. Brückner, J. Org. Chem. 2004, 69 , 5116-5119.
19 A. Hassannia, G. Piercy, N. Merbouh, Lett. Org. Chem. 2009, 6, 478-480.
20 P. P. Pradhan, J. M. Bobbitt, W. F. Bailey, Org. Lett. 2006, 8, 5485-5487.
21 T. Miyazawa, T. Endo, Tetrahedron Lett. 1986, 27 , 3395-3398.
22 P. P. Pradhan, J. M. Bobbitt, W. F. Bailey, J. Org. Chem. 2009, 74 , 9524-9527.
23 T. Endo, T. Miyazawa, S. Shiihashi, M. Okawara, J. Am. Chem. Soc. 1984, 106, 3877-3878.
24 N. Merbouh, J. M. Bobbitt, C. Brückner, Tetrahedron Lett. 2001, 42 , 8793-8796.
25 A. L. Bartelson, J. M. Bobbitt, W. F. Bailey, 2009, Unpublished material.
26 A. D. Findlay, A. Gebert, I. A. Cade, M. G. Banwell, Aust. J. Chem. 2009, 62 , 1173-1180.
27 V. A. Golubev, V. N. Borislavskii, A. L. Aleksandrov, Russ. Chem. Bull. 1977, 9, 1874-1881. (English translation)
28 M . F . S e m m e l h a c k , C . R . S c h m i d , D . A . C o r t é s , Tetrahedron Lett. 1986, 27 , 1119-1122.
29 W. F. Bailey, J. M. Bobbitt, K. B. Wiberg, J. Org. Chem. 2007, 72 , 4504-4509.
30 L. O. Atovmyan, V. A. Golubev, N. I. Golovina, G. A. Klitskaya, J. Struct. Chem. 1975, 16 , 79-83. (English translation)
31 T. Takata, Y. Tsujino, S. Nakanishi, K. Nakamura, E. Yoshida, T. Endo, Chem. Lett. 1999, 937-938.
32 V. A. Golubev, E. V. Kobylyanskii, J. Org. Chem. USSR 1972, 8, 2657-2662. (English translation)
33 V. A. Golubev, T. S. Rudyk, V. D. Sen’, A. L. Aleksandrov, Russ. Chem. Bull. 1976, 25, 744-750. (English translation)
34 V. A. Golubev, é. G. Rozantsev, M. B. Neiman, Russ. Chem. Bull. 1965, 14, 1898-1904. (English translation)
35 Y. Liu, H. Guo, Z. Liu, Huaxue Xuebao 1991, 49 , 187-192. (in Chinese)
36 M. Yamaguchi, T. Takata, T. Endo, Tetrahedron Lett. 1988, 29 , 5671-5672.
37 T. Miyazawa, T. Endo, M. Okawara, J. Org. Chem. 1985, 50, 5389-5391.
38 J. M. Bobbitt, Unpublished work.
39 T. Miyazawa, T. Endo, Yuki Gosei Kagaku Kyokaishi (J. Synth. Org. Chem. Jpn.) 1986 , 44 , 1134-1144. (in Japanese)
40 J. M. Bobbitt, Z. Ma, D. Bolz, T. Osa, Y. Kashiwagi, Y. Yanagisawa, F. Kurashima, J. Anzai, J. E. Tacorante-Morales, in Oxoammonium salt oxidations of alcohols in the presence of, and on the surfaces of, an electrode, an ion exchange resin, and silica gel, London, UK, 1998.
41 D. M. Philipp, R. Muller, W. A. Goddard, K. A. Abboud, M. J. Mullins, R. V. Snelgrove, P. S. Athey, Tetrahedron Lett. 2004, 45, 5441-5444.
42 A. Abad, C. Agulló, A. C. Cunat, R. H. Perni, Tetrahedron: Asymmetry 2000, 11, 1607-1615.
43 T. V. Ovaska, J. A. Sullivan, S. I. Ovaska, J. B. Winegrad, J. D. Fair, Org. Lett. 2009, 11 , 2715-2718.
44 T. R. Hoye, M. Hu, J. Am. Chem. Soc. 2003, 125, 9576-9577.
45 J. Hudon, T. A. Cernak, J. A. Ashenhurst, J. L. Gleason, Angew. Chem. Int. Ed. 2008, 47 , 8885-8888.
46 D. M. Troast, J. Yuan, J. A. Porco Jr, Adv. Synth. Catal. 2008, 350, 1701-1711.
47 K. A. B. Austin, M. G. Banwell, G. J. Harfoot, A. C. Willis, Tetrahedron Lett. 2006, 47 , 7381-7384.
48 P. T. Gulyas, S. J. Langford, N. R. Lokan, M. G. Ranasinghe, M. N. Paddon-Row, J. Org. Chem. 1997, 62 , 3038-3039.
49 L. W. Habel, S. De Keersmaecker, J. Wahlen, P. A. Jacobs, D. E. De Vos, Tetrahedron Lett. 2004, 45 , 4057-4059.
50 D. H. Hunter, J. S. Racok, A. W. Rey, Y. Z. Ponce, J. Am. Chem. Soc. 1988, 53 , 1278-1281.
51 J. M. Bobbitt, Z. Ma, Heterocycles 1992, 33 , 641-648.
52 V. A. Golubev, R. I. Zhdanov, I. T. Protsishin, é. G. Rozantsev, Russ. Chem. Bull. 1970, 19, 2043. (English translation)
53 V. A. Golubev, R. V. Miklyush, J. Org. Chem. USSR 1972, 8, 1376-1377. (English translation).
54 Y. Liu, T. Ren, Q. Guo, Chin. J. Chem. 1996, 14 , 252-258.
55 S. Weik, G. Nicholson, G. Jung, J. Rademann, Angew. Chem. Int. Ed. 2001, 40 , 1436-1439.
56 Z. Ma, Ph.D. Dissertation, University of Connecticut, Storrs, CT, 1991.
57 M. Sch?mann, H. J. Sch?fer, Electrochim. Acta 2005, 50 , 4956-4972.
58 T. Breton, D. Liaigre, E. M. Belgsir, Tetrahedron Lett. 2005, 46 , 2487-2490.
59 J. M. Bobbitt, M. C. F. Guttermuth, Z. Ma, H. Tang, Heterocycles 1990, 30 , 1131-1140.
60 D. H. Hunter, D. H. R. Barton, W. J. Motherwell, Tetrahedron Lett. 1984, 25 , 603-606.
61 H.-X. Guo, Y.-C. Liu, Z.-L. Liu, C.-L. Li, Res. Chem. Intermed. 1992, 17 , 137.
62 Y. B. Ding, L. Yang, Z. H. Liu, Y. C. Liu, J. Chem. Res., Synop. 1994, 328-329.
63 Y. Liu, W. Wang, Q. Guo, Chin. Chem. Lett. 1996, 7, 790-793.
64 P. Pradhan, J. M. Bobbitt, Unpublished work.
注:本文為提供者整理翻譯的,由于知識所限,錯誤在所難免,敬請原諒。如有問題可以查找原文。
快速導航
化學品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
關于物競
物競數據庫是一個全面、專業、專注,并且免費的中文化學品信息庫,為學生、學者、化學品研究機構、檢測機構、化學品工作者提供專業的化學品平臺進行交流。
數據庫采用全中文化服務,完全突破了中英文在化學物質命名、化學品俗名、學名等方面的差異,所提供的數據全部中文化,更方便國內從事化學、化工、材料、生物、環境等化學相關行業的工作人員查詢使用。
關注我們

-
微信賬號:物競化學品數據庫

-
微博賬號:wjhxp
聯系我們
上海市延長路149號上海大學科技園412室
公司總機: 021-56389801
訂購電話: 4007001514
傳真電話: 021-56389802
客服電話: 021-56332350
電子郵件: wingch@basechem.org
 滬公網安備 31010602001115號
滬公網安備 31010602001115號