從維他命B12依賴性酶中得到的環境友好型催化劑
- 2012-12-10
- 專題
1.介紹
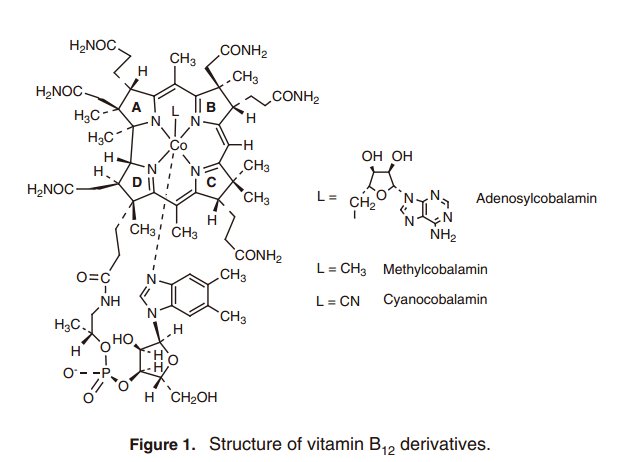
眾所周知維他命B12衍生物是一種獨特的輔酶,它內部具有鈷-碳(Co–C)鍵,與多種脫輔基蛋白組合參與各種催化功能。1) 維他命B12是一個金屬配合物,在咕啉環中鈷離子與四個氮原子環相互配合(圖1)。它最初用作治療惡性貧血的神奇武器,它獨特的結構是由Hodgkin等人通過X-ray crystallography所描述。2)中央鈷離子的氧化價態由+1到+3; Co(I)呈灰綠色, Co(II)呈黃色-橙色,Co(III),是最常見的形式,在B12衍生物中顯紅色,也被稱為“紅色維生素。”甲鈷胺和腺苷鈷胺作為體內酶輔酶形式在生物合成蛋白酸酶和碳骨架異構化重排中分別具有催化功能。3) 最近的研究表明,例如咕啉環復合物結構在厭氧細菌的脫氯反應中起到活性中心的作用;這表明B12依賴酶的一個新的功能(方案1)。4)
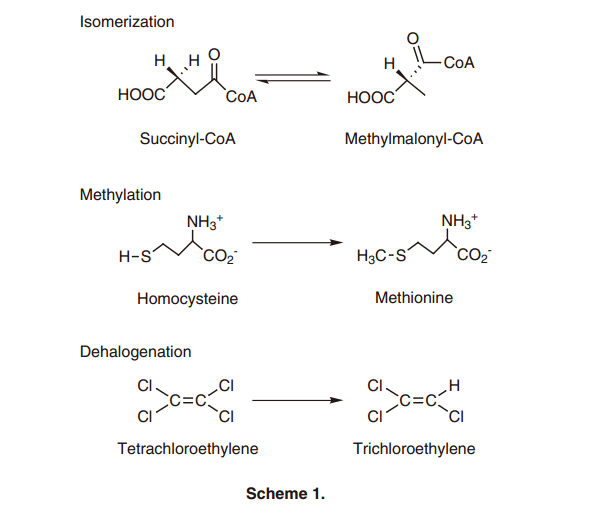
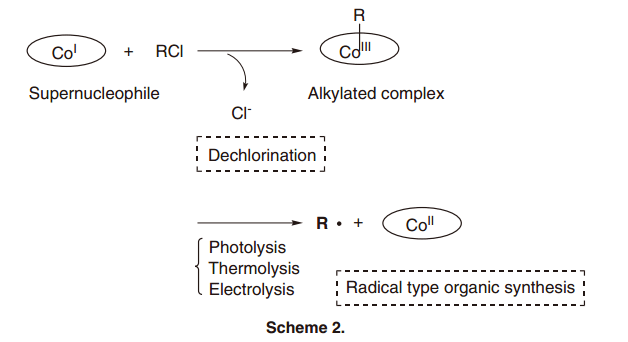
B12依賴酶反應的顯著特點是與金屬碳鍵形成了一個獨特的有機金屬結構。這種結構主要是由Co(I)與有機鹵化物,Co(III)與Grignard試劑反應所得, 它很容易在溫和的外源刺激,比如說可見光、熱、氧化還原條件下發生均裂。特別是, 當前反應發生時,Co-C鍵的形成包含了有機鹵化物的脫鹵,并產生有機自由基種在的Co-C鍵的斷裂(方案2)。通過專注于上述Co-C鍵的特點, 我們研發了一個分子轉換,利用Co-C鍵與B12模型復合物的形成于分裂來激化B12依賴酶的活性中心。5)在這篇文章中, 我們提出了我們的結果:維他命B12衍生物的合成及其催化過程。
2. 維生素B12建模
Vitamin B12從氰鈷胺生產生物體重大量獲得,用于膳食補充或者是牲畜飼養。目前為止,沒有任何關于使用天然維生素B12作為催化劑原料的安全性或者是經濟型方面的問題產生。然而, 從細菌中獲得的天然維生素B12可能回含有維生素B12相關物質,這表明它有可能會具有較差的化學穩定性。這不是因為咕啉環的結構的不穩定性,而是側鏈取代基的退化。正如后面鎖描述的, 咕啉環具有很高的穩定性。要使用vitamin B12衍生物作為催化劑, 應滿足下列條件:(1)應保留咕啉環結構,(2)中央鈷原子應該是完好的, (3)修改的側鏈應該維持不變。在這三個條件中, 我們暫時從氰鈷胺中移除生理必須的側鏈結構,通過所有的外周側鏈的化學修飾作用生成酯用于合成疏水性維生素B12(heptametyl cobyrinate) (圖2, A).6) 酸催化劑參與下,通過加熱醇溶液中的氰鈷胺的簡單合成,可以獲得80%的疏水性vitamin B12,即使氰鈷胺被用作原料,它也是通過以上提到的大量的微生物所得到的。使用疏水性維生素B12作為起始原料,它可以合成各種B12催化劑,并且也具有廣泛的應用(圖2, B-D)。天然維生素B12的咕啉環仍然存在于上述配合物種,因此中心鈷離子合成B12衍生物的氧化還原潛力和電子屬性類似于天然B12,這表明合成B12 衍生物在分子轉換中顯示出像有機酶一樣的高反應性。6)

3. 使用B12配合物的有機電反應
如上所述,B12衍生物的親核Co(I)與有機鹵化物反應,在生成的烷基化合物的同時隨后脫鹵反應。因此,它很可能會發展成為一種活性Co(I)類形成烷基化配合物的方法,這是一個B12依賴酶反應的關鍵中間體。合成的B12配合物可以通過化學還原試劑比如硼氫化鈉、金屬鋅、鈉汞齊等還原, 因此大量使用這些化學試劑是不符合綠色化學的。因此電化學技術被用于生成B12配合物Co(I)類,在這個反應中,B12配合物作為電反應中介物(圖3)。7)
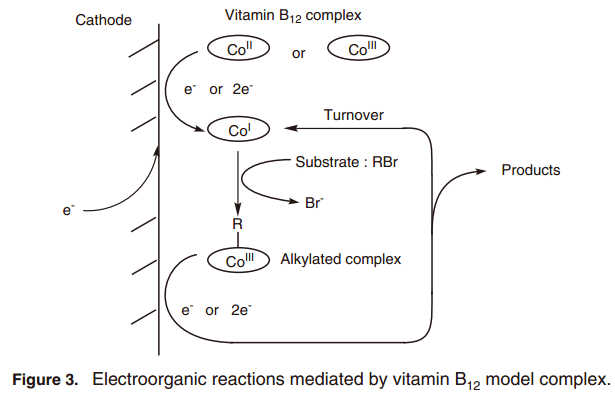
我們通過控制電位電勢在-1.4或者-1.5Vvs來開發各種分子變換. Ag/AgCl利用疏水性維生素B12作為一個中介物(方案3)。例如, 當溴代丙烯酸酯作為基質,通過烷基配合物的均裂分子內環化有機自由基合成大圓環內酯(方案3, eq. 1).8)中間體烷基化配合物形成利用電噴霧電離質譜(ESI-MS)和紫外-可見光譜通過檢測電解溶液被直接觀測到。疏水性維生素B12也能有效地充當環境污染物的脫氯催化劑,例如如二氯二苯基三氯乙烷(DDT)(方案3, eq. 2). 9)值得注意的是,在反應過程中,由于氯原子脫氯處理成無害的氯離子,B12的催化體系不產生有毒二次污染物如氯氣和光氣(方案2)。此外, B12催化劑反應后不會降解。它的高耐用性在反應過后通過UV-vis和質譜分析后確認。當其他的鈷配合物,如卟啉配合物,在相同條件下,電解使用過后復合物嚴重退化。
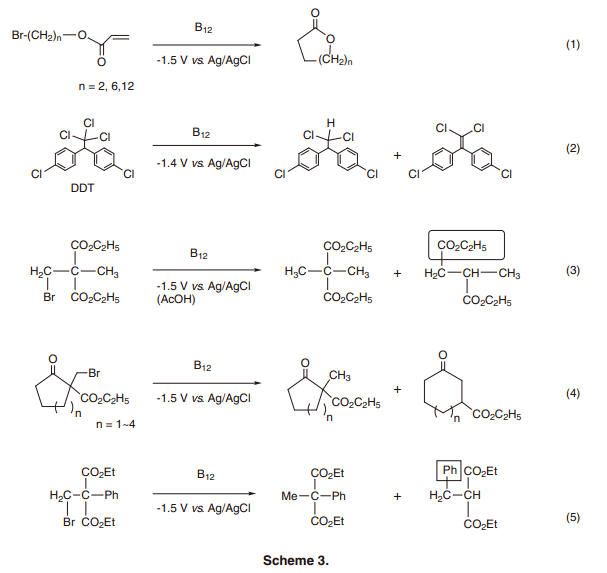
為了重復使用這個強大的B12催化劑, B12配合物被固定在電極上或者溶解在離子溶液中用于試驗過后回收或者循環使用。在前者的情況下,B12配合物被固定在鉑電極的表面上,通過引入一個三甲氧基硅烷基側鏈,覆蓋率約在1.6×10–10mol/cm2(2, B)。使用苯乙級溴作為模板的反應性能進行了評估,證明反應活性很高,6,000周轉數/h(圖4)。10) 反應結束后,產物和B12催化劑可以很容易的進行分離,因為催化劑被固定在電極上面。其他的B12修飾電極也有了報道,比如說聚合物覆蓋電極,界面聚合電極,和溶膠-凝膠的FLM摻雜電極。11-13)
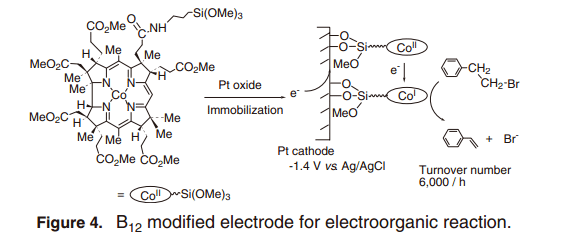
另一方面,當離子液體作為反應溶劑,B12配合物作為離子溶液,顯示出以下各種好處。一般情況下,有機電反應在含有電解質的極性有機溶劑中反應。然而,使用含有多種電解質的有機溶劑的這種反應相對于當前節約使用化學試劑的理念是不可取的,即使是該程序被用于降解氯化有機化合物。最近, 離子溶液中有機電反應被越來越多的報道用于解決這些問題。離子液體在通常壓力下是室溫液體鹽,其特點是由它的不易揮發性,防火阻燃,和優異的導電性,并且是優異的有機電反應溶劑。14)離子溶液中使用B12配合物作為催化劑,DDT電解發生,同樣的方式,含有支持電解質的極性溶劑中脫氯反應同樣的有效的進行。此外,反應結束后的產品和B12配合物的提取在有機層和離子液體層中分別提取。反應中溶于離子液體的B12配合物可以回收再用(圖5)。15) 更有趣的是, 離子液體中B12配合物的反應性提高,即在二甲基甲酰胺(DMF)反應中,基板的轉換效率被提高了四倍。這種在DMF的離子液體增強的反應可以被解釋為休斯英戈爾德預測中的溶劑的極性對反應速率的影響。16) CO(I)與DDT產生的電反應是一種“Menschutkin type of reaction”其中兩個中性反應物Co(I) 和DDT, 反應形成的帶電產品在極性離子液體中通過電荷分離的活化絡合物,這最終降低激活能量,導致反應速率的增加。類似的促進效果反應在對-硝基苯磺酸甲酯與三-n-丁胺離子液體中唄密切觀測到。17)其實,離子液體中ETN value18) 已經被確認為極性指數。例如, 1-正丁基-3 -甲基咪唑四氟硼酸鹽的指數為0.673, 表明它是高極性的極性溶劑,例如DMF (ETN= 0.386). 19)
4. B12配合物的光敏性反應
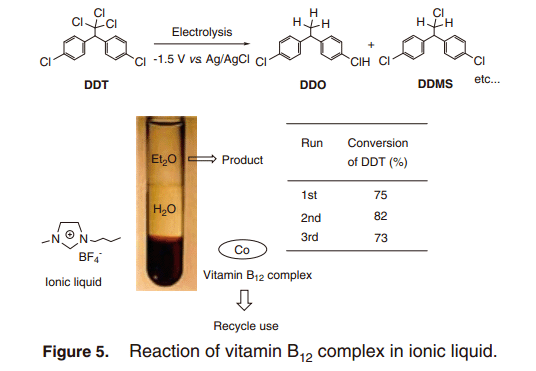
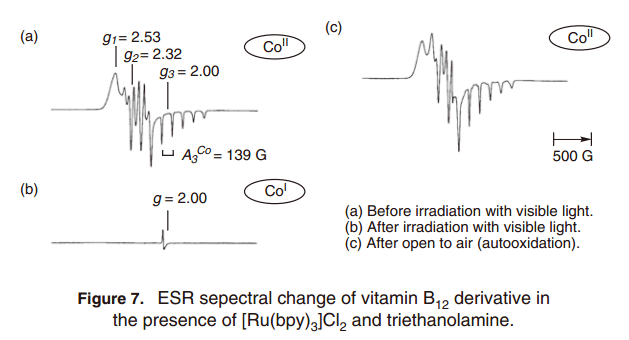
我們試圖設計一個光驅動催系統,可以利用B12 配合物開發一個清晰的分子轉換。光是地球上豐富,清潔的能源之一,并已用于在有機合成中的很長一段時間。因此,我們注意到了三聯吡啶配合物釕(II)([RuII(bpy)3]Cl2), 一直廣泛的用作光敏劑,并使用的光誘導電子轉移構建了催化系統用于鈷(I)類的制造反應。20) [RuII(bpy)3]Cl2配合物在可見光照射下活躍,且[Ru(bpy)3]+ 配合物具有很高的還原電勢(–1.35 V vs. Ag/AgCl) ,通過犧牲電子給體三乙醇胺還原淬火得到。21) 因此,它能夠由從釕光敏劑的電子轉移產生催化活性B12 配合物(E1/2(CoII/CoI) =–0.6Vvs. Ag/AgCl)。如圖6所示, 在釕光敏劑和犧牲給電子供體參與下,利用B12配合物DDT的脫氯反應發生。因此, 三小時內大部分DDT 被轉換為DDD, 一個單脫氯產品。在沒有B12配合物或者是黑暗條件下反應幾乎沒有進展。因此,人們認為Co(I)是通過光誘導電子轉換反應產生,可以作為引發反應的活性體。ESR譜更改證實,釕光敏劑電子轉移反應還原了B12配合物生成鈷(I)的物種。ESR特征信號用于檢測B12順磁性Co(II),而相應的ESR信號用于光反應中反磁性Co(I)(圖7).
5. B12-TiO2混合催化劑
最近,由金屬絡合物和光敏材料半導體組成的混合催化劑已經有所報道。22-23)根據該技術,很有可能開發出一種金屬絡合物與半導體互為效果的混合催化劑。因此,我們專注于二氧化鈦,24)這是一個n-type半導體,用于開發一個光源驅動B12-TiO2混合催化劑。氧化鈦(TiO2)-傳導帶電子能量的降低(–0.5 V vs.正常氫電極(NHE)) 25) 使B12 絡合物還原成具有催化活性的Co(I) species, 這表明光能驅動混合催化劑可以通過固定B12絡合物于TiO2上開發合成。在這種情況下,二氧化鈦不僅起到了B12絡合物的腳手架的作用,但還擔任還原B12絡合物的電子源(圖8)。這里,通過多個維生素B12的羧基(圖2,C型)和TiO2表面羥基基團的相互作用將B12絡合物固定在TiO2上。約3-4×10-5摩爾(40-50毫克)的維生素B12絡合物(cobyrinic酸)被發現固定在1克二氧化鈦上面,70%-80%的TiO2表面覆蓋著B12絡合物。B12絡合物被牢固打的固定在TiO2表面上,保持穩定1年以上,甚至后期分散于各種有機溶劑,如醇,并不通過超聲波處理分離。

當氯化有機化合物比如說DDT的降解反應, 利用此混合催化劑進行反應時,僅僅幾毫克的催化劑便可在一天時間里激活DDT使得脫氯反應100倍作用。26)混合催化劑通過光能激活,因此化學試劑或者昂貴的反應器都不需要。不可見光(365 nm) 所提供的紫外線照射為激發TiO2提供足夠的能量。此外, B12絡合物和TiO2都是無毒的,因此B12-TiO2混合催化劑可能是環境/人類友好型催化劑。另外, 我們研究有機合成中使用混合催化劑的自由基反應,該結果表明這種混合催化劑可應用于各種分子轉換,包括上述環擴反應(方案3,式4)和1,2-遷移的官能團(方案3,式5)。因此,混合催化劑作為一種替代,可以使用tin化合物(Bu3SnH/AIBN system)用于常規的自由基合成試劑。
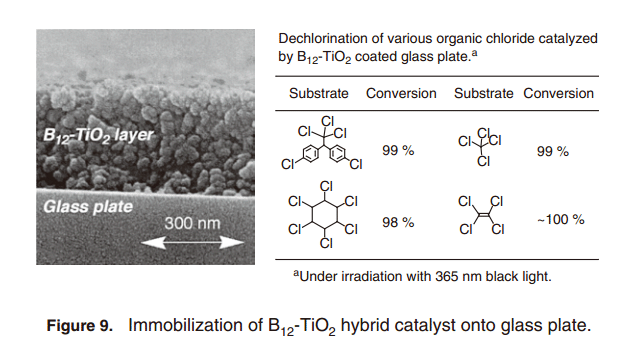
當它被轉換成漿料溶膠溶液的時候,二氧化鈦也可以被固定在載體上,如玻璃基板和珠子上面。TiO2薄膜(約幾百納米厚度), 通過在玻璃基質上浸涂得到,通過浸入到含羧基集團的B12絡合物溶液,與B12 絡合物進行雜交反應。因此,在玻璃板上面制備B12-TiO2混合催化劑也可以催化上述反應(圖9)。玻璃板面上固定B12-TiO2混合催化劑可以簡化反應過程,并且,以便于從反應混合物中分離產物。
6. 維生素B12-超支化聚合物復合催化劑
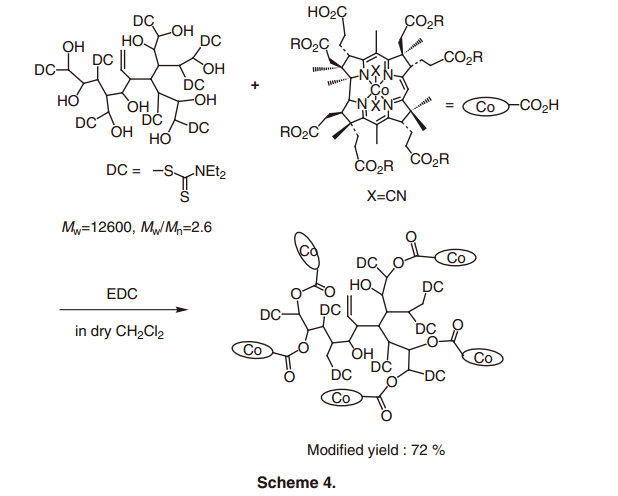
超支化聚合物是樹突狀聚合物打的一種,通過簡單經濟的方法單步驟合成,與通過多步聚合反應合成的樹枝狀聚合物相比,它具有各種優點。27)此外,與線性聚合物相比,該聚合物具有較高的溶解性和較低的溶液粘度。它預期可以使用在一個均勻的溶液中。28)事實上, 超支化聚合物,由于其高度支化結構有許多末端官能團,適合攜帶官能分子。29)考慮到由超支化聚合物提供的納米空間作為一個新的反應領域,B12-超支化聚合物復合催化劑得以合成(方案4)。30) 所使用的維生素B12配合物的量可以在超支化聚合物的末端官能團調整到某處之間的幾%和70%,并且可以容易地控制密度的B12配合物。當B12配合物密度確定,相鄰的復合物的合作效果便于苯乙基溴的二聚化反應。31)
7. 結語
我們概述了從B12依賴酶到分子轉換的發展過程。混合性催化酶包含金屬配合物的合成,與B12酶的活性中心相似,激活各種分子變換,包括環境友好的有機合成反應和有機鹵化物污染物的降解反應的有機電反應或光化學反應。我們推出的這種仿生化學的發展將對下一代的科學和技術起到重要的作用。
參考文獻
1.(a) Vitamin B12 and B12-Proteins ed. by B. Kr?utler, D. Arigoni, B. T. Golding, Wiley-VCH 1998; (b) Chemistry and Biochemistry of B12 ed. by R. Banerjee, Wiley-Interscience 1999.
2.(a) D. C. Hodgkin, J. Pickworth, J. H. Robertson, K. N. Trueblood, R. J. Prosen, J. G. White, Nature 1955, 176, 325-328; (b) D. C. Hodgkin, J. Kamper, M. Mackay, J. Pickworth, K. N. Trueblood, J. G. White, Nature 1956, 178, 64-66; (c) P. G. Lenhert and D. C. Hodgkin, Nature 1961, 192, 937-938.
3.(a) R. Banerjee and S. W. Ragsdale, Annu. Rev. Biochem. 2003, 72, 209-247; (b) T. Toraya, Chem. Rev. 2003, 103, 2095-2127; (c) B. Kr?utler, Biochem. Soc. Trans. 2005, 33, 806-810.
4.(a) G. Glod, U. Brodmann, W. Angst, C. Holliger, R. P. Schwarzenbach, Environ. Sci. Technol. 1997, 31, 3154-3160; (b) C. Holliger, G. Wohlfarth, G. Diekert, FEMS Microbiol. Rev. 1999, 22, 383-398; (c) B. Kr?utler, W. Fieber, S. Ostermann, M. Fasching, K. –H. Ongania, K. Gruber, C. Kratky, C. Mikl, A. Siebert, G. Diekert, Helv. Chim. Acta 2003, 86, 3698-3716; (d) S. Ruppe, A. Neumann, G. Diekert, W. Vetter, Environ. Sci. Technol. 2004, 38, 3063-3067; (e) K. M. McCauley, D. A. Pratt, S. R. Wilson, J. Shey, T. J. Burkey, W. A. van der Donk, J. Am. Chem. Soc. 2005, 127, 1126-1136; (f) J. E. Argüello, C. Costentin, S. Griveau, J. –M. Sáveant, J. Am. Chem. Soc. 2005, 127, 5049-5055.
5.Reviews: (a) Y. Hisaeda, T. Nishioka, Y. Inoue, K. Asada, T. Hayashi, Coord. Chem. Rev. 2000, 198, 21-25; (b) K. L. Brown, Chem. Rev. 2005, 105, 2075-2150.
6.(a) Y. Murakami, Y. Hisaeda, A. Kajihara, Bull. Chem. Soc. Jpn. 1983, 56, 3642-3646; (b) Y. Murakami, Y. Hisaeda, A. Kajihara, T. Ohno, Bull. Chem. Soc. Jpn. 1984, 57, 405-411.
7.R. Scheffold, G. Rytz, L. Walder, Modern Synthetic Methods, Vol. 3, ed. By R. Scheffold, John Wiley & Sons, 1983, 355-440.
8.H. Shimakoshi, A. Nakazato, T. Hayashi, Y. Tachi, Y. Naruta, Y. Hisaeda, J. Electroanal. Chem. 2001, 507, 170-176.
9.(a) H. Shimakoshi , M. Tokunaga, Y. Hisaeda, Dalton Trans. 2004, 878-882; (b) H. Shimakoshi, Y. Maeyama, T. Kaieda, T. Matsuo, E. Matsui, Y. Naruta, Y. Hisaeda, Bull. Chem. Soc. Jpn. 2005, 78, 859-863.
10.H. Shimakoshi , M. Tokunaga, K. Kuroiwa, K. Kimizuka, Y. Hisaeda, Chem. Commun. 2004, 50-51.
11.(a) A Ruhe, L. Walder, R. Scheffold, Helv. Chim. Acta 1985, 68, 1301-1311; (b) Y. Murakami, Y. Hisaeda, T. Ozaki, Y. Matsuda, J. Chem. Soc., Chem. Commun. 1989, 1094-1096; (c) D.-L. Zhou, C. K. Njue, J. F. Rusling, J. Am. Chem. Soc. 1999, 121, 2909-2914; (d) C. K. Njue and J. F. Rusling, Electrochem. Commun. 2002, 4, 340-343.
12.R. Fraga, J. P. Correia, R. Keese, L. M. Abrantes, Electrochim. Acta 2005, 50, 1653-1655.
13.H. Shimakoshi, A. Nakazato, M. Tokunaga, K. Katagiri, K. Ariga, J. Kikuchi, Y. Hisaeda, Dalton Trans. 2003, 2308-2312.
14.T. Welton, Chem. Rev. 1999, 99, 2071-2083.
15.M. Jabbar, H. Shimakoshi, Y. Hisaeda, Chem. Commun. 2007, 1653-1654.
16.K. A. Cooper, M. L. Dhar, E. D. Hughes, C. K. Ingold, B. J. MacNulty, L. I. Woolf, J. Chem. Soc. 1948, 2043-2046.
17.L. Crowhurst, N. L. Lancaster, J. M. P. Arlandis, T. Welton, J. Am. Chem. Soc. 2004, 126, 11549-11555.
18.C. Reichardt, Solvent and Solvent Effects in Organic Chemistry, VCH (UK) Ltd., Cambridge, UK, 2nd edn, 1998.
19.(a) S. N. V. K. Aki, J. F. Brennecke, A. Samanta, Chem. Commun. 2001, 413-414; (b) M. J. Muldoon, C. M. Gordon, I. R. Dunkin, J. Chem. Soc., Perkin Trans. 2, 2001, 433-435.
20.(a) H. Shimakoshi , M. Tokunaga, T. Baba, Y. Hisaeda, Chem. Commun. 2004, 1806-1807; (b) H. Shimakoshi, S. Kudo, Y. Hisaeda, Chem. Lett. 2005, 34, 1806-1807.
21.A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser, A. V. Zelewsky, Coord. Chem. Rev. 1988, 84, 85-277.
22.(a) S. O. Obare, T. Ito, G. J. Meyer, Environ. Sci. Technol. 2005, 39, 6266-6272; (b) Y. Astuti, E. Palomarse, S. A. Haque, J. R. Durrant, J. Am. Chem. Soc. 2005, 127, 15120-15126; (c) S. O. Obare, T. Ito, G. J. Meyer, J. Am. Chem. Soc. 2006, 128, 712-713; (d) B. I. Ipe and C. M. Niemeyer, Angew. Chem. Int. Ed. Engl. 2006, 45, 504-507.
23. R. J. Kuhler, G. A. Santo, T. R. Caudi l l, E. A. Betterton, R. G. Arnold, Environ. Sci. Technol. 1993, 27, 2104-2111.
24.(a) M. A. Fox, Acc. Chem. Res. 1983, 16, 314-321; (b) M. D. Ward, J. R. White, A. J. Bard, J. Am. Chem. Soc. 1983, 105, 27-31; (c) M. A. Fox and M. T. Dulay, Chem. Rev. 1993, 93, 341-357; (d) M. R. Hoffmann, S. T. Martin, W. Choi, D. W. Bahnemann, Chem. Rev. 1995, 95, 69-96.
25.A. Fujishima, T. N. Rao, D. A. Tryk, J. Photochem. Photobiol. C: Photochem. Rev. 2000, 1, 1-21.
26.H. Shimakoshi, E. Sakumori, K. Kaneko, Y. Hisaeda, Chem. Lett. 2009, 38, 468-469.
27.M. Jikei and M. Kakimoto, Prog. Polym. Sci. 2001, 26, 1233-1285.
28.P. H. Toy and K. D. Janda, Acc. Chem. Res. 2000, 33, 546-554.
29.(a) K. Ishizu and A. Mori, Polym. Int. 2001, 50, 906-910; (b) K. Ishizu, T. Shibuya, A. Mori, Polym. Int. 2002, 51, 424-428; (c) K. Ishizu, T. Shibuya, J. Park, S. Uchida, Polym. Int. 2004, 53, 259-265.
30.K. Tahara, H. Shimakoshi, A. Tanaka, Y. Hisaeda, Tetrahedron Lett. 2007, 48, 5065-5068.
31.K. Tahara, H. Shimakoshi, A. Tanaka, Y. Hisaeda, unpublished result.
注:本文為提供者整理翻譯的,由于知識所限,錯誤在所難免,敬請原諒。如有問題可以查找原文。
快速導航
化學品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
關于物競
物競數據庫是一個全面、專業、專注,并且免費的中文化學品信息庫,為學生、學者、化學品研究機構、檢測機構、化學品工作者提供專業的化學品平臺進行交流。
數據庫采用全中文化服務,完全突破了中英文在化學物質命名、化學品俗名、學名等方面的差異,所提供的數據全部中文化,更方便國內從事化學、化工、材料、生物、環境等化學相關行業的工作人員查詢使用。
關注我們

-
微信賬號:物競化學品數據庫

-
微博賬號:wjhxp
聯系我們
上海市延長路149號上海大學科技園412室
公司總機: 021-56389801
訂購電話: 4007001514
傳真電話: 021-56389802
客服電話: 021-56332350
電子郵件: wingch@basechem.org
 滬公網安備 31010602001115號
滬公網安備 31010602001115號