烯類單體在室溫離子液體中的自由基聚合反應研究進展
- 2012-12-07
- 專題
室溫離子液體是一類在室溫(或低于100℃)呈熔融狀態的鹽, 通常由體積較大的有機陽離子和無機酸根離子組成[1,2]。與常見的有機溶劑相比, 有如下一些特點:(1)蒸汽壓低、揮發性弱, 除了能滿足一般化學反應和分離提純操作的需要外, 還適用于要求高真空或較高溫度的反應體系;(2)穩定性好, 不易燃、易爆;(3)溶解能力易調, 容易回收利用;(4)極性較高、絡合能力較弱, 是使用過渡金屬催化劑反應的理想介質;(5)對某些反應有一定的催化和選擇作用。基于這些獨特的性質, 室溫離子液體被認為是繼超臨界CO2之后的新一代“綠色”溶劑, 相關研究十分活躍。
關于離子液體的合成、性質和應用, Seddon[1]、Welton[2]等已有詳細的評介。自由基聚合反應涉及的聚合物量大、面廣, 本文將主要介紹烯類單體在室溫離子液體中自由基聚合反應的研究情況, 并結合筆者正在進行研究進展, 提出對今后工作的粗淺看法。
1室溫離子液體中的自由基聚合反應
1. 1普通自由基聚合反應
Hong等[3,4]以偶氮二異丁腈(AIBN)和過氧化苯甲酰(BPO)為引發劑, 分別研究了甲基丙烯酸甲酯(MMA)和苯乙烯(St)在1-丁基-3-甲基咪唑六氟磷酸鹽([bmim][PF6])和苯中的自由基聚合反應。發現在離子液體中聚合得到的聚甲基丙烯酸甲酯(PMMA)和聚苯乙烯(PSt)與在苯中得到的相應聚合物具有相似的分子量分布, 但在離子液體中的聚合速度快, 聚合物分子量比在苯中聚合得到的大約10倍。他們認為主要原因可能有二:一是聚合過程中聚合物析出, 增長鏈末端的自由基由于被高分子鏈包埋而減少了鏈轉移和終止的機會;另一是離子液體的粘度比苯高, 增長鏈自由基通過擴散會聚而發生碰撞的幾率比在苯中的小。這兩個原因都可使自由基的壽命延長, 聚合速度增加。但后來的研究表明, 雖然PSt在[bmim][PF6]中不溶, 但PMMA 在[bmim][PF6]中是可溶的[5~7],MMA在[bmim][PF6]中聚合時反應體系一直保持均相。因此, 前述的第一個原因不能解釋MMA在[bmim][PF6]中聚合速度快、聚合物分子量大的事實。
Harrisson等[5~6]采用脈沖激光聚合(pulsed laser polymeriztion)技術深入地研究了MMA在離子液體中鏈增長速率常數(kp)與離子液體濃度的關系。結果表明, kp隨離子液體濃度的提高而增加, 最高可超過本體中的2倍, 遠遠高于一般有機溶劑對kp的影響。雖然尚不完全清楚為什么離子液體具有這么明顯的溶劑誘導加速聚合作用, 但可部分解釋為什么MMA在[bmim][PF6 ]中聚合速度快、聚合物分子量大。
普通自由基聚合反應一般不能用來合成嵌段共聚物。利用離子液體中增長鏈自由基壽命較長的特點, Zhang等[8]通過順序加料的方法, 用自由基聚合制備了聚苯乙烯和聚甲基丙烯酸甲酯嵌段聚合物。使用的離子液體是[bmim] [PF6],引發劑是BPO, 反應溫度為70℃。先合成PSt鏈段, 聚合到一定程度后減壓將未反應的St抽出, 再加入第二單體MMA于室溫聚合, 即可得到PSt-b-PMMA 嵌段共聚物, 并經1HNMR、DSC和分級沉淀等方法證實。
溶劑的回收利用是所有溶液聚合都必須面對的問題, 因為殘存的溶劑不僅會造成環境污染, 而且會嚴重影響高分子制品的最終性能。離子液體蒸氣壓低、不易揮發, 不能用常見的加熱或抽真空的方法除去, 必須采用其它的方法。Benton等[9]研究了離子液體的回收利用問題。聚合完成后, 用V(乙醇)/V(水)= 80/20的混合溶劑萃取含有[bmim][PF6]的PMMA 薄膜。在23℃時, 如果時間足夠長, 最終有97%的離子液體被萃取出來; 而在55℃時, 則萃取速度要快得多, 2h內可萃取出87%的離子液體。旋干萃取液, 即可回收離子液體。
殘留的少量離子液體可讓其保留在聚合物中, 起增塑劑的作用。Scott等[10]發現[bmim] [PF6]是PMMA 的有效增塑劑。當聚合物中離子液體的重量濃度達到50%時仍然是均相體系, 而當常用的增塑劑鄰苯二甲酸二辛酯的濃度達到40%~50%時已有明顯的相分離發生。此外, 以該離子液體為增塑劑的聚合物力學性能與用鄰苯二甲酸二辛酯為增塑劑的相當, 但熱穩定性更好。
1. 2活性自由基聚合反應
1. 2. 1ATRP聚合反應 原子轉移自由基聚合反應(ATRP)是迅速發展的一種活性自由基聚合技術[11], 由有機鹵化物(引發劑)、過渡金屬低價態鹽(催化劑)及含氮多齒配體構成引發體系, 反應機理如圖1所示。聚合過程中, 低價金屬離子從引發劑分子奪取一個鹵原子后變成高價金屬離子,而引發劑分子失去鹵原子后生成自由基, 該自由基向單體加成誘導聚合反應。增長鏈自由基也可以從高價態過渡金屬鹽奪取一個鹵原子后失去活性, 生成休眠種; 同時, 將金屬鹽從高價態還原為低價態。引發劑的“活化”與“失活”是可逆反應, 速度很快, 反應體系中活性中心的濃度遠較普通自由基聚合體系中的低, 并且保持恒定, 從而有效調控聚合反應的進行。
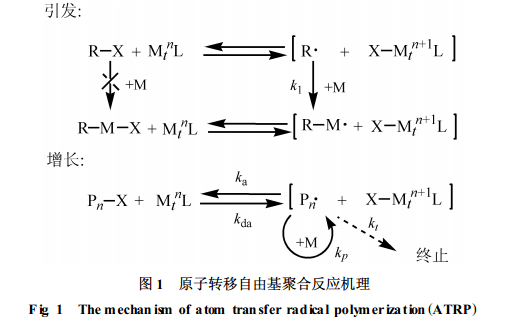
從上述的反應機理可以看出, 反應體系中過渡金屬化合物的含量對于聚合反應的控制起著決定性的作用。普通有機溶劑對過渡金屬化合物的溶解性差, 在其中進行的ATRP一般都是非均相反應。為了達到對聚合反應有效控制的目的, 往往需要加入較大量的金屬催化劑, 這一方面增加了反應成本, 另一方面又不可避免地造成殘留金屬離子對聚合物的污染。為了解決這—問題,Carmichael等[7]開展了MMA在[bmim][PF6]中進行ATRP反應的嘗試, 如圖2所示。所用的引發劑是2-溴異丁酸乙酯, 催化劑是CuBr, 有機配體是N-丙基-2-吡啶基甲亞胺(N-proyl-2-pyridylmethanimine)。該聚合反應具有反應速度快、反應溫度低(低至30℃)、聚合物分子量分布窄的特點。由于[bmim][PF6 ]與甲苯不相溶, 反應完成后可用甲苯從反應混合物中將聚合物萃取出來, 而絕大多數金屬離子仍留在離子液體中, 不會污染聚合物。

Sarbu等[12,13]合成了一系列陽離子為1-丁基-3-甲基咪唑鎓、陰離子分別為Cl-、Br-、A lCl4-、CO32-、(C4H9)2PO4-的離子液體, 系統地研究了陰離子對MMA在其中的ATRP反應的影響。當以2-溴異丁酸乙酯為引發劑, 在催化劑量的離子液體存在下,MMA在本體、甲苯或苯甲醚等溶劑中可以進行ATRP反應。如果以Fe(Ⅱ)為催化劑, 不使用配體也可調控MMA的活性聚合反應;而如果以Cu(Ⅰ)為催化劑, 則只有當[bmim][(C4H9)2PO4]存在時才可不使用配體。含有過渡金屬鹽的離子液體很容易與非極性的聚合物溶液分離, 催化劑的回收和循環使用也可以方便地解決。
Biedron等[14]報道了一系列丙烯酸酯單體在[bmim][PF6]中的ATRP聚合反應。單體在[bmim][PF6]中的溶解性取決于單體分子上烷基的長度。丙烯酸甲酯(MA)在[bmim] [PF6]中的反應為均相反應, 所得聚合物分子量的實測值與計算值接近, 且分子量分布較窄。丙烯酸丁酯(BA)雖然不溶于[bmim][PF6],為非均相聚合反應, 但當攪拌速度足夠快能夠保證引發劑、單體和增長鏈自由基在兩相之間的轉移時, 聚合反應仍以可控的方式進行。而對丙烯酸己酯和丙烯酸十二烷基酯, 即使施以充分攪拌, 反應仍然不可控。
基于前面的工作,Biedron等[15]又研究了丙烯酸酯類單體在離子液體中的嵌段共聚合反應。在[bmim][PF6]中, 先用ATRP方法合成聚丙烯酸丁酯大分子引發劑, 在BA幾乎完全聚合后, 直接加入第二種單體MA, 所得嵌段聚合物分子量分布很窄, 均聚物含量很少, 數均分子量比理論值稍高。反之, 如果先在離子液體中合成PMA 作為大分子引發劑, 在MA轉化率低于70%時加入第二單體BA, 仍可得到分布較窄的嵌段聚合物。但是, 當轉化率高于70%時再加BA, 聚合產物中除了嵌段共聚物外還有均聚物。而當轉化率大于90%時再加BA, 則根本得不到嵌段共聚物。端基分析表明, 在均相體系中, 當MA的轉化率高于70%時, 增長鏈自由基會發生不可逆終止; 而在非均相體系中, 增長鏈自由基不可逆終止的速率要小得多。
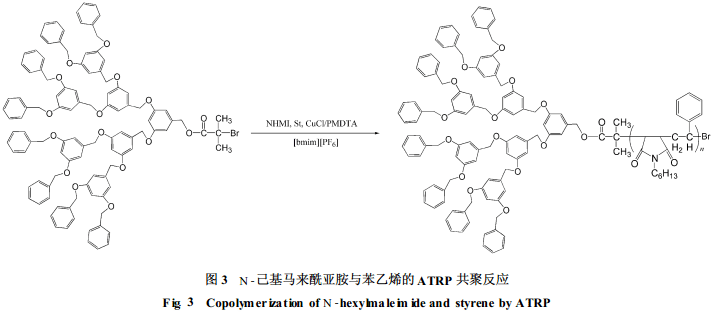
習復等[16]以含溴端基、不同代數的樹枝狀分子Dendriticpolyacrylether 2-bromoisobutyra te為引發劑, 用ATRP方法合成了N-己基馬來酰亞胺與苯乙烯共聚物, 如圖3所示。該反應的引發效率很高(99%), 反應溫度低(30℃), 所得聚合物的分子量分布較窄(1.18<Mw/Mn<1.36), 受催化劑的污染較少。與在普通有機溶劑如苯甲醚中進行的反應相比, 在較大的投料比范圍內, 在離子液體中更易形成N-己基馬來酰亞胺和苯乙烯的交替共聚物。
1.2.2 反向ATRP聚合反應
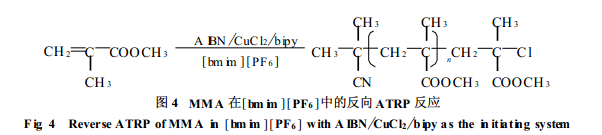
反向ATRP與ATRP一樣, 也是借助過渡金屬催化劑調控的氧化還原反應降低反應體系中增長鏈自由基的濃度, 實現對聚合反應的控制。在ATRP中, 引發劑是烷基鹵化物, 催化劑是過渡金屬的低價鹽。前者對人體和環境的危害較大, 后者使用時易被氧化。為了克服ATRP的這兩個缺點,Matyiaszewski等[17]提出了反向ATRP的概念。其主要特點是用AIBN、BPO等普通自由基聚合引發劑代替烷基鹵化物, 用過渡金屬高價鹽代替低價鹽, 從而有效解決了普通ATRP中引發劑的毒性和催化劑的易氧化性問題。
筆者研究了MMA在[bmim][PF6]中的反向ATRP反應, 采用的引發體系是AIBN/CuC l2/bipy(聯吡啶), 如圖4所示。Matyjaszewski等曾報道AIBN/CuCl2/bipy引發體系不能有效調控MMA的自由基聚合反應, 主要原因是催化劑在本體中或其它有機溶劑中溶解性較差, 為非均相聚合體系。同時, 單體MMA的反應活活較高, 使體系中自由基濃度過高而無法控制。而當以4, 4’-二(5-壬基)-2, 2’-聯吡啶(dNbipy)為配體、苯甲醚為溶劑形成均相體系時, 反應則可控[18]。筆者利用離子液體對于金屬化合物良好的溶解性, 選用[bmim][PF6]作為聚合反應的溶劑, 可形成均相反向ATRP聚合體系[19]。通過對不同溫度下聚合反應動力學的研究以及對聚合物端基的分析, 確定該聚合反應為活性聚合, 所得聚合物的分子量分布很窄(Mw/Mn<1.2)。另外, 由于催化劑在[bmim][PF6]中的良好溶解性, 只需加入少量催化劑([AIBN][CuCl2]= 8/1)即可有效調控聚合反應, 從而減少了金屬離子對聚合物的污染。從反應混合物中分離聚合物后, 僅需經過簡單處理, 離子液體即可回收并重復使用。再向回收的離子液體中加入MMA和AIBN, 反應又可以同樣的方式進行。
以上工作是在疏水性離子液體中完成的, 筆者還進行了MMA在親水性離子液體[C4mim][BF4]和[C12mim][BF4]中的反向ATRP反應[20]。文獻報道含BF4-的離子液體對過渡金屬化合物有更好的溶解性[21], 因而希望在其中的自由基聚合反應可以得到更好的控制。為了進一步研究聚合時手性環境對聚合物立構規整性的影響, 筆者設計合成了一類含有不對稱中心的離子液體[22], 化學結構如下:
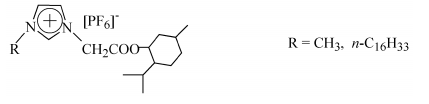
由于離子液體對催化體系具有很好的溶解性, 僅向反應體系中加入少量的手性離子液體m(MMA)/m(ILs)=3/1即可調控MMA的反向ATRP聚合反應, 獲得分子量分布很窄的聚合物(Mw/Mn<1.2)。以所得的聚合物作為大分子引發劑, 可引發甲基丙烯酸薄荷醇酯(MnMA)的ATRP反應, 獲得具有光學活性的嵌段聚合物。
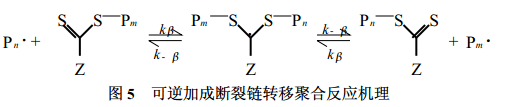
1. 2. 3 RA F T聚合反應 可逆加成斷裂鏈轉移聚合(RAFT)是新近發展起來、適用單體范圍更廣的一種活性聚合技術, 其反應機理如圖5所示[23]。與普通自由基聚合反應相比, 體系中除了單體、引發劑外, 還有調控增長鏈自由基濃度的二硫代羰基化合物。聚合時, 引發劑引發單體聚合形成增長鏈自由基, 增長鏈自由基向二硫代羰基化合物的CS 鍵可逆加成形成自由基中間體, 該中間體可逆斷裂又形成新的增長鏈自由基。由于在反應過程中, 自由基的濃度遠比二硫代羰基化合物的濃度低, 鏈終止和鏈轉移的程度降低, 從而可達到控制聚合反應的目的。
Perrier等研究了AIBN引發的MMA、MA和St在離子液體中的RAFT聚合反應[24]。他們選用的二硫代羰基化合物和離子液體的化學結構為:
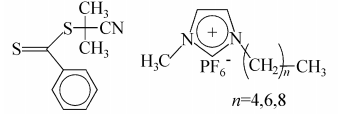
聚苯乙烯不溶于所用的離子液體, 在反應的初期就沉淀出來, 基本得不到聚合物。MMA、MA及其聚合物在三種液體中都有很好的溶解性, 聚合以活性的方式進行, 所得聚合物的分子量分布(PDI)較窄。對PMMA, PD I值小于1. 1; 而對PMA, PDI值雖然比PMMA 的大, 但仍小于1.3。在離子液體中的聚合速度比在本體和其它溶劑中的大, 并且和離子液體的結構有關。在其它條件完全相同的情況下,M M A在[C4mim] [PF6]中的轉化率是84. 3% ,而在[C6mim] [PF6]和[C8mim] [PF6]中的轉化率分別為91.3%和90.1%。所得聚合物的末端基確定, 可與新加入的單體進行擴鏈反應。
2 展望
Seddon[25]曾指出, 幾乎所有類型的有機反應都可以在離子液體中進行。現在, 大量的實驗事實已經證明了他的預言。離子液體所提供的反應環境與傳統的溶劑有很大差別, 從而產生了許多新反應、新現象、新結構和新功能, 并且有可能減少污染, 保護環境。可以說, 離子液體為化學科學和化學工業的發展提供了新的機遇。相比之下, 離子液體中烯類單體的自由基聚合反應研究開展得還很少, 有許多尚待探索的領域, 今后的工作可能包括新型離子液體的設計、合成、表征以及離子液體中的自由基聚合反應規律、活性自由基聚合反應、立構規整自由基聚合反應等方面。其中, 功能性離子液體的設計、合成及在烯類單體自由基聚合反應中的應用將受到進一步的關注。功能性離子液體是指既可作為反應介質, 又具有催化、分子識別、絡合等功能的離子液體, 如離子液晶、手性離子液體、帶有配位基團的離子液體等。它們可以提供不同于一般有機溶劑的反應環境, 從而有助于在更高水平上實現對聚合反應和高分子結構的控制。此外, 利用離子液體合成其它類型的高分子已有一些報道, 毫無疑問這也將是今后發展的重要方向。
參 考 文 獻
[1] KRSeddon. J. Chem. , T ech. B io techno l. , 1997, 68: 351~356.
[2] TW elton. Chem. R ev. , 1999, 99: 2071~2083.
[3] HWZhang, KLHong, J MM ay s. Po lym. P rep. , 2001, 42: 583~584.
[4] KLHong, HWZhang, J MM ay seta l. Chem. Comm un. , 2002, 13: 1368~1369.
[5] SH a rrisson, SRM ackenze, DMH add leton. Po lym. P rep. , 2002, 43: 883~884.
[6] SH a rrisson, SRM ackenzie, DMH add leton. Chem. Comm un. , 2002, 23: 2850~2851.
[7] AJ Ca rm ichael, DMH add leton, SAFBoneta l. Chem. Comm un. , 2000, 14: 1237~1238.
[8] HWZhang, KLHong, J WM ay s. M acrom o lecu les, 2002, 35: 5738~5741.
[9] MGB en ton, CSB razel. Po lym. P rep. , 2002, 43: 881~882.
[10] MPSco t t, CSB razel, MGB en toneta l. Chem. Comm un. , 2002, 13: 1370~1371.
[11] J SW ang, KM a ty ja szew sk i. J. Am. Chem. Soc. , 1995, 117: 5614~5615.
[12] TSa rbu, KM a ty ja szew sk i. Po lym. P rep. , 2002, 43: 211~212.
[13] TSa rbu, KM a ty ja szew sk i. M acrom o l. Chem. Phy s. , 2001, 202: 3379~3391.
[14] TB ied ro, PKub isa. M acrom o l. R ap idComm un. , 2001, 22: 1237~1242.
[15] TB ied ro, PKub isa. J. Po lym. Sci. Po lym. Chem. , 2002, 40: 2799~2809.
[16] YL Zhao, JMZhang, J J iang eta l. J. Po lym. Sci. Po lym. Chem. , 2002, 40: 3360~3366.
[17] J SW ang, KM a ty ja szew sk i. M acrom o lecu les, 1995, 28: 7572~7573.
[18] J HX ia, KM a ty ja szew sk i. M acrom o lecu les, 1997, 30: 7692~7696.
[19] HYM a, XHW an, XFCheneta l. J. Po lym. Sci. Po lym. Chem. , 2003, 40: 143~151.
[20] HYM a, XHW an, XFCheneta l. Po lym er, 2003, 44: 5311~5316.
[21] LCB ranco, J NRo sa, J JMR am o seta l. Chem2Eu r. J. , 2002, 8: 3671~3677.
[22] HYM a, XHW an, XFCheneta l. Ch ineseJ. Po lym. Sci. , 2003, 21: 265~270.
[23] J Ch iefa ri, YKChong, FE rco leeta l. M acrom o lecu les, 1998, 31: 5559~5562.
[24] SPerrier, TPD avis, AJ Ca rm ichaeleta l. Chem. Comm un. , 2002, 19: 2226~2227.
[25] MF reem an t le. C&EN. , 1999, 4: 23~24.
注:本文為提供者整理翻譯的,由于知識所限,錯誤在所難免,敬請原諒。如有問題可以查找原文。
快速導航
化學品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
關于物競
物競數據庫是一個全面、專業、專注,并且免費的中文化學品信息庫,為學生、學者、化學品研究機構、檢測機構、化學品工作者提供專業的化學品平臺進行交流。
數據庫采用全中文化服務,完全突破了中英文在化學物質命名、化學品俗名、學名等方面的差異,所提供的數據全部中文化,更方便國內從事化學、化工、材料、生物、環境等化學相關行業的工作人員查詢使用。
關注我們

-
微信賬號:物競化學品數據庫

-
微博賬號:wjhxp
聯系我們
上海市延長路149號上海大學科技園412室
公司總機: 021-56389801
訂購電話: 4007001514
傳真電話: 021-56389802
客服電話: 021-56332350
電子郵件: wingch@basechem.org
 滬公網安備 31010602001115號
滬公網安備 31010602001115號