逆流色譜分離手性化合物
- 2012-12-07
- 專題
手性化合物是自然界生命體中普遍存在的物質,其在醫藥、農藥、香料等眾多領域內起著非常重要的作用。為了滿足對光學純化合物的需要,各國科學家都在尋找有效、便捷的拆分手性化合物的技術[1],近年來逆流色譜在分離手性化合物上顯示出了獨特的優勢。用逆流色譜分離手性化合物的主要優點是:逆流色譜是一種不用固態支撐體或載體的液液分配技術,在分離過程中完全消除了氣、液色譜中常見的不可逆吸附現象,不會破壞分子,適用于分離極性大的手性化合物以及生物大分子[2];由于它獨特的分離機制,使其可用于制備分離,一次最大可以分離近 10g 的樣品[3]。逆流色譜技術主要可以分為液滴逆流色譜、旋轉腔室逆流色譜以及離心分配逆流色譜三個大類,本文主要介紹這些逆流色譜的工作原理及其在分離手性化合物方面的應用。
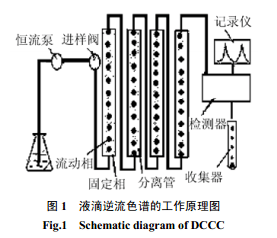
1 液滴逆流色譜(DCCC)
液滴逆流色譜儀(droplet countercurrent chromatography) 是Ito最早研發出來的[4]。它主要是通過在縱向排列的分離管柱里,用互不相溶的兩相中的下相作固定相,將其先充滿管柱,另一相作移動相,帶著樣品從管柱的下面往上連續通過分離管,由于重力的作用,移動相會在固定相中形成小液滴,樣品將在移動相和固定相之間有效地進行分配。由于分配系數的不同,使得不同組分在分離柱中向前移動的速度不同,經過不斷的分配傳遞之后,樣品組份將按分配系數的大小順序從管系的出口流出,從而達到分離的目的。圖1表示出了DCCC儀器的裝置示意圖。
DCCC的分離效率完全取決于液滴的形成狀況,只有當上升的小液滴具有最佳尺寸,剛好占據管柱的斷面時,才能獲得最好的分離效果。所以溶劑的選擇很重要,常見的二元和三元溶劑系統有:己烷/水,氯仿/水,己烷/甲醇/水,氯仿/甲醇/水。
1984年Takeuchi等[5]從Davanoov等[6]合成N-正十二烷基脯氨酸(C12-Pro)中受到啟發,使用當時流行的液滴逆流色譜儀(含400根直徑為2cm的分離管,總容積為2L),用2mmol/L合成的正十二烷基-L-脯氨酸作手性添加劑,2mmol/L的正丁醇和 1mmol/L 的醋酸銅-醋酸緩沖溶液作溶劑系統,移動相流速保持1.1mL/min,花了2.5d 的時間,對亮氨酸對映異構體進行了基線分離,同時部分拆開了(±)纈草氨酸和(±)蛋氨酸。
兩年后,Oya等[7]用(-)-R-2-氨基丁醇作手性添加劑,拆分了100mg 的二環[2.2.1]-庚-5-烯-2-羧酸衍生物,比傳統的結晶法和酯化法拆分對映異構體的效果更好。所用儀器的柱容積是280mL,溶劑系統為氯仿/甲醇/磷酸鹽緩沖溶液pH=7(7/13/8),耗時2.3d。由于液滴逆流色譜僅靠流動相在重力作用下形成液滴,洗脫速率很難提高,所以耗時較長。而且單靠在分離管中簡單地上升或者下降的移動相所帶來的溶質在兩相中的反復分配是很有限的,分離效率也不高。而旋轉腔室逆流色譜的發展在一定程度上彌補了液滴逆流色譜的不足。
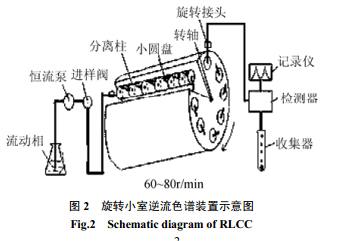
2 旋轉腔室逆流色譜(RLCC)
旋轉腔室逆流色譜(rotation locular countercurrent chromatography) 是由多根裝置在轉軸周圍的玻璃分離管柱組成,在管內應用聚四氟乙烯的小圓盤將空柱分成多個小腔室,這些小腔室少則幾十個,多則可上百個。小圓盤的中心有一個直徑大約為1mm的小穿孔,其作用是可以讓流動相通過分離柱,當樣品帶朝向相鄰的橫向擴散時又會受到腔室間小穿孔的限制,這種機制能形成較高的分配效率。當移動相帶著樣品通過小腔室里面兩相溶劑間的界面時,由于管柱在重力場中的旋轉使樣品帶在每個腔室里面兩相之間平緩而更有效地分離分配。與液滴逆流色譜相比,它大大增加了兩相溶劑系統的相互作用,也增加了溶質在兩相中的分配次數,從而提高了分離效果。該方法不要求流動相在固定相中一定能形成小的液滴,所以其大大擴展了流動相的選擇范圍,也拓展了該法的應用領域。
圖2 是旋轉小室逆流色譜裝置示意圖。
1982年Hostettman 等[8]用旋轉腔室逆流色譜儀,首次在逆流色譜中使用R,R-酒石酸-二-5-壬基酯為手性添加劑[9],用1,2-二氯乙烷/水作溶劑系統拆分了200mg(±)旋光異構體麻黃堿。在分離過程中,0.3mmol/L手性試劑被加在作為移動相的有機相中,水相中加入0.5mmol/L 六氟磷酸鈉緩沖溶液,樣品用水相溶解,移動相流速為17~20mL/min,儀器旋轉速度為60~70r/min,實驗溫度為2~3℃和5~8℃,一次進樣花了3~4d,向人們展示了利用逆流色譜實現對旋光異構體分離的良好潛力。雖然旋轉腔室逆流色譜分離效果比DCCC有所提高,但所需時間仍然較長,分離量不大。
3 離心分配逆流色譜(CPC)
離心分配逆流色譜(centrifugal partition countercurrent chromatography)是逆流色譜中內容最豐富的,各種不同的設備類型多達數十種。離心逆流色譜有兩種基本的原理:流體動力學平衡體系(HDES)和流體靜力學平衡體系(HSES)[10]。根據這些原理制造的儀器對應分為兩個大類:行星式逆流色譜和非行星式逆流色譜。因它們都是具有離心力作用的兩相溶劑系統,故又稱為離心分配逆流色譜。
兩相溶劑體系的選擇對于這類逆流色譜成功分離樣品十分關鍵。兩相溶劑系統應滿足以下要求[11]:(1)為了保證固定相保留值合適(不低于50%),溶劑體系的分層時間須小于30s;(2)手性樣品在兩相中的分配系數 K 最好在1左右;(3)盡量采用揮發性溶劑,以方便后續處理,易于樣品純化。
3.1 非行星式逆流色譜
這類逆流色譜儀與流體動力學平衡體系的逆流色譜不同,分離管柱固定在分離儀中,只公轉,不自轉。在眾多的這類色譜中,目前真正還在得到應用的主要是匣盒式離心逆流色譜。匣盒式分離柱實際上是由一些小分離管組成,它們分布在離心轉軸的周圍,其縱軸與離心力平行,它們之間用聚四氟乙烯小管串聯,工作時先泵入固定相,分離柱轉動時,泵入移動相,移動相帶著樣品在分離柱中形成一連串液滴通過固定相,樣品進行連續分配分離[12]。
Shinomiya先使用RLCC,用牛血清蛋白(BSA) 作手性添加劑拆分D,L-犬尿氨酸[13],花了60h仍沒有得到基線分離,后改用離心分配色譜,溶劑系統10(wt)% PEG800作固定相,5(wt)% 的磷酸二氫鈉緩沖溶液和6(wt)% BSA作移動相,轉速800r/min,流速0.2mL/min,只耗時 3.5h 就成功拆分2.5mg D,L-色氨酸[14],說明CPC 的分離能力比RLCC更強。
3.2 行星式逆流色譜
在行星式逆流色譜儀中,分離柱采用了除了繞中心軸旋轉外本身還作自轉運動的模式。這種運動能造成兩相溶劑反復劇烈混合和分層,其分配轉移頻率達到13次/s,使溶質達到比較理想的分離分配,因而用較少的溶劑就能快速有效地分離樣品[15]。
Duret在用行星式逆流色譜儀(13mL 容積)進行手性拆分實驗時,選用甲苯和水作溶劑系統,把在液相[16]、薄層色譜[17]、毛細管電泳[18]中有效的手性添加劑萬古霉素借用過來,使水中溶解140mg/mL萬古霉素,調整pH=4.7,拆分D,L-丹磺酰正亮氨酸。分離過程中,當從尾到頭泵入移動相時,左消旋體被洗脫出來,當相反泵入移動相時,右消旋體被洗脫出來。實驗表明用萬古霉素也適用于作逆流色譜的手性添加劑,而且最大能分離50mg 的D,L-丹磺酰正亮氨酸[19]。
此種設備中最具有代表性的商業化儀器是高速逆流色譜儀(HSCCC),我國絕大多數的研究、應用以及國產儀器的生產也主要集中在該類型的色譜上,現予以專門詳述。
4 高速逆流色譜
高速逆流色譜(High-speed countercurrent chromatography)是Ito 研發的新一代逆流色譜。目前國內研制的高速逆流色譜(HSCCC)儀器緊緊同國際最新發展相配合。該裝置核心部件分離柱由多層聚四氟乙烯管繞成的線圈構成,多個線圈成對稱分布或用平衡器平衡,因分離柱線圈旋轉快,固定相保留多,兩相作用充分,分離效果好,分離速度快,正被廣泛用在眾多領域的分離分析,在分離手性化合物上也得到了應用。圖3即為高速逆流色譜儀裝置示意圖。

脯氨酸的衍生物是較早使用的逆流色譜手性添加劑,因為它們能提供π電子,易于對具有相似電荷特征的手性化合物進行拆分[20]。Pirkle等[21]使用高速逆流色譜,由這類手性添加劑在庚烷/乙酸乙酯/甲醇/水(體積比3/1/3/1)溶劑系統中較快(80min)拆開了兩種DNB-氨基酸衍生物。隨后,Ito等[22]使用這類具有π電子的手性添加劑,同樣使用HSCCC,選用了兩種溶劑系統:正己烷/乙酸乙酯/甲醇/10mmol/L 的鹽酸(體積比 8/2/5/5)和正己烷/乙酸乙酯/甲醇/10mmol/L 鹽酸(體積比6/4/5/5),成功分離了(±)DNB-苯甘酸、(±)DNB-苯丙酸等[23],通過改變手性添加劑在固定相中的量和濃度,一次進樣最多能分離1g 量的手性樣品[24,25]。
隨著逆流色譜技術的逐步完善,分離能力的不斷提高,使它能越來越多的借鑒其它色譜技術上的有效手性添加劑,這方面最好的例子是磺化-β-環糊精在高速逆流色譜上的成功應用。磺化-β-環糊精首先用在毛細管電色譜[26]上很成功地分離了7-去甲基-奧美昔芬(DMO)。由于磺化-β-環糊精已是商業化的產品,如果能用在逆流色譜上獲得制備性分離,將有較大的應用前景。研究人員隨后在高速逆流色譜上做了大量探索實驗,最終選用乙酸乙酯/甲醇/水(體積比10/1/9)作溶劑系統,水相中含2%的磺化-β-環糊精作為手性添加劑,基線分離了(±)7-DMO[27]。為逆流色譜手性添加劑的選擇打開了思路。
目前,筆者課題組主要進行高速逆流色譜儀分離手性藥物的研究,如用羧甲基環糊精作手性添加劑對氨魯米特和撲爾敏的外消旋體進行了制備性分離,用樟腦磺酸作手性添加劑對阿司咪唑和拉貝洛爾外消旋體進行了制備性分離,下面是在最佳色譜條件下分離撲爾敏外消旋體的色譜圖,其最佳溶劑系統為:乙酸乙酯/甲醇/水(體積比10/1/9) ,最適合的手性添加劑濃度為20mmol/L[28]。

5 pH 區帶精制逆流色譜
pH區帶精制逆流色譜是上世紀90年代初期由Ito[29]提出后逐漸發展起來的,屬于高速逆流色譜中的一項特殊技術,目前的文獻報道都是在一般的高速逆流色譜儀上完成的。如在少量酸性物質的樣品溶液中加入一定濃度的有機酸時,可以產生一個非同尋常的窄而尖的峰,當樣品量增大時,每個組分將在柱中形成一個高度濃縮的等 pH 區帶,并以一個矩形峰被洗脫出來,當樣品量繼續增大,矩形峰也隨著增寬,但并不影響組分之間的分離效果,這就使該方法能在一般高速逆流色譜法的基礎上成十倍地增加樣品進樣量,而不用對常見設備作任何改進。方法操作簡單、易行、分離量大,有工業化應用的前景。圖5示出了pH區帶精制逆流色譜中的尖峰、矩形峰和 pH突躍。
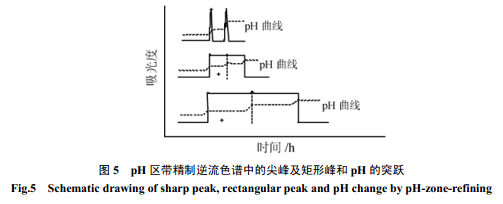
pH區帶精制逆流色譜技術是利用樣品的 pKa 值和分配系數的不同進行質子交換來提純樣品的。它的分析分離可用下式表示:pH=pKa+log{KD/D(1+[CS]K±)-1},K±是(+)、(-)對映異構體與手性添加劑形成復合物的穩定常數,[CS]為在固定相中手性添加劑的濃度,(+)、(-)對映異構體的 pKa、KD是相同的,D是保留值,通過復合物的K+和K-的不同及手性添加劑在固定相中的濃度不同使對映異構體分開[27]。Oliveros等[20]用三氟乙酸作固定相、氨在水中作移動相,用330mL 容積的高速逆流色譜儀,經過 3h 分離了2g 的(±)DNB-亮氨酸[21]。pH區帶提取逆流色譜已經成為分離量最大的制備分離手性化合物的逆流色譜技術。
隨著越來越多的多糖、生物堿、氨基酸、抗生素等作為手性固定相在氣液相色譜中不斷獲得成功[30],一些研究人員把尋找有效的逆流色譜手性添加劑的目光投到了天然產物和它們的衍生物上。當Linder 等[31]發現從金雞納樹皮中提取的奎寧(QN)和奎寧定(QD)是一種有效的離子交換色譜的手性添加劑后,又在此基礎上把奎寧和奎寧定衍生化為更強立體選擇性的手性添加劑金剛烷基氨基甲酰基QN-Selector 1和金剛烷基氨基甲酰基QD-Selector 2,它們易于接受和失去質子,在分離氨基酸時立體選擇性很好。圖6為奎寧及其衍生化的手性添加劑的化學結構式。

通過使用高速逆流色譜,Franco 等[32]用0.1mmol/L pH6.0的氨基乙酸緩沖液/三戊基乙醇/甲醇/庚烷/( 體積比 10/5/1/5)作溶劑系統,含10.6mmol/L的手性添加劑,轉速為1000r/min,流速3mL/min,很好地分離了DNB-亮氨酸、DNZ-新戊基甘氨酸和 DNZ-β-苯丙氨酸。用0.1mol/L pH8.0的氨基乙酸緩沖液/丙酮/甲基異丁基酮(MIBK)(體積比2/1/2)最大分離了300mg的DNB-亮氨酸。用pH區帶精制逆流色譜,含三氟乙酸(10mmol/L)和QN-Selector 1 的MIBK溶液作固定相,含20mmol/L的氨水作移動相,轉速為1200r/min,流速3mL/min,最大分離了1.2g的DNB-亮氨酸。
6 結語
目前利用逆流色譜分離手性化合物的論文還不多,制約著手性分離的主要因素是能否找到合適的高選擇性手性添加劑。根據逆流色譜分離手性化合物的報道,從氣、液、毛細管電泳中廣泛使用的手性添加劑中,找到適合于逆流色譜的手性添加劑,仍然是一個很好的途徑。隨著高效手性添加劑的發現和逆流色譜儀器的進一步更新完善,逆流色譜制備性分離手性化合物的能力將會進一步提高,關于這方面的研究和應用也將會進一步加深和擴大。
參考文獻
[1] N M Maier, P Franco, W Lindner. J. Chromatogr. A, 2001, 906: 3~33.
[2] A P Foucault, L Chevolot. J. Chromatogr. A, 1998, 808: 322.
[3] A Marston, I Slacanin, K Hostettmann. Physochemical Analysis, 1990, 1: 3~17.
[4] N K Hostettman. Planta Medica, 1980, 39: 1~18.
[5] V A Davankov, A S Bochkov, Y P Belov. J. Chromatogr., 1981, 218: 547~557.
[6] T Takeuchi, R Horikawa, T Tanimura. J. Chromatogr., 1984, 284: 285~288.
[7] S Oya, J K Snyder. J. Chromatogr., 1986, 370: 333~338.
[8] B Domon, K Hostettmann, K Kovacevic et al. J. Chromatogr., 1982, 250:149~151.
[9] V Prelog, Z Stojanac, K Kovacevic. Helv. Chim. Acta., 1982, 65: 377~384.
[101] A Berthod, D W Armstrong. J. Liq. Chromatogr., 1988, 11: 547~583.
[11] Y Ito, W D Conway . High-speed Counter-current Chromatography, Chemical Analysis. New York:Wiley, 1996: 121~175.
[12] Y Ito. Crc. Crit. Rev. Anal. Chem., 1986, 17: 65~143.
[13] F Oka, H Oka, Y Ito. J. Chromatogr., 1991, 538: 99~108.
[14] T Arai, H Kuroda. Chromatographia , 1991, 32: 56.
[15] D W Armstrong, Y Tang, S Chen et al. Anal. Chem., 1994, 66: 1473~1484.
[16] D W Armstrong, Y Zhou. J. Liq. Chromatogr., 1994, 17: 1695~1707.
[17] D W Armstrong, K Rundlett , G L Reid III . Anal. Chem., 1994, 66: 1690~1695.
[18] P Duret , A Foucault, R Margraff . J. Liq. Chromatogr, 2000, 23: 295~312.
[19] Y Ito, M Weinstein, I Aoki , R Harada et al. Nature, 1966, 212: 985.
[20] L Oliveros, P P Franco, C Minguillon et al. J. Liq. Chromatogr., 1994, 17: 2301~2318.
[21] W H Pirkle, P G Murray. J. Chromatogr., 1993, 641: 11~19.
[22] Y Ma, Y Ito, A Foucault. J. Chromatogr. A, 1995, 704: 75~81.
[23] Y Ma, Y Ito. Anal. Chem., 1995, 67: 3069~3074.
[24] YMa, Y Ito. Anal. Chem., 1996, 68: 1207~1211.
[25] Y Ma, Y Ito, A Berthod. J. Liq. Chromatogr., 1999, 22: 2945~2955.
[26] J Breinholt, S V Lehmann, A R Varming. Chirallity, 1999, 11: 768~771.
[27] A P Foucault. J. Chromatogr., 2001, 906: 365~378.
[28] L M Yuan. J C H Liu. Z H Yan et al. J. Liq. Chromatogr., in press.
[29] A Weisz , A L Scher, K Shinomiya et al. J. Am. Chem. Soc., 1994, 116: 704~708.
[30] G Subramarian. Chiral Separations Techniques. A Practcical Approach. 2nd ed.; Weinheim: Wiley-VCH, 2001. 46.
[31] W Lindner, M Lammerhofer, N M Maier. PCT/EP: 97/02888, 1997.
[32] P Franco, J Blanc, W R Oberleitner et al. Anal. Chem., 2002, 74: 4175~4183.
注:本文為提供者整理翻譯的,由于知識所限,錯誤在所難免,敬請原諒。如有問題可以查找原文。
快速導航
化學品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
關于物競
物競數據庫是一個全面、專業、專注,并且免費的中文化學品信息庫,為學生、學者、化學品研究機構、檢測機構、化學品工作者提供專業的化學品平臺進行交流。
數據庫采用全中文化服務,完全突破了中英文在化學物質命名、化學品俗名、學名等方面的差異,所提供的數據全部中文化,更方便國內從事化學、化工、材料、生物、環境等化學相關行業的工作人員查詢使用。
關注我們

-
微信賬號:物競化學品數據庫

-
微博賬號:wjhxp
聯系我們
上海市延長路149號上海大學科技園412室
公司總機: 021-56389801
訂購電話: 4007001514
傳真電話: 021-56389802
客服電話: 021-56332350
電子郵件: wingch@basechem.org
 滬公網安備 31010602001115號
滬公網安備 31010602001115號